
|
|
Производство стерильной продукции
25. Стерильные радиофармацевтические препараты разделяются на две группы: - препараты, подлежащие финишной стерилизации, т.е. стерилизации в первичной окончательной упаковке; - препараты, не допускающие финишной стерилизации, производство которых должно быть организовано в асептических условиях. Требования к чистоте помещений зависят от того, к какой группе относится препарат. Требования к чистоте рабочих зон, в которых продукт или первичная упаковка может находиться в контакте с окружающим воздухом, приведены в Приложении 1 к настоящему стандарту. 26. Для определения требований к перепадам давления, направлению потока воздуха и его качества могут использоваться методы анализа рисков. 27. В закрытых автоматизированных системах, представляющих собой, как правило, горячие камеры с размещением в них установок химического синтеза, систем очистки, стерилизующей фильтрации "на линии", должна быть предусмотрена зона C. В горячие камеры, находящиеся в закрытом состоянии, должен подаваться воздух с высокой степенью чистоты. Асептические операции должны выполняться в зоне A. 28. До начала производства сборка стерильного оборудования и компонентов (трубок, стерилизующих фильтров), укупорка и герметизация стерильных флаконов должны выполняться в асептических условиях.
Документация
29. Порядок разработки, пересмотра, утверждения и выдачи документов, относящихся к производству радиофармацевтических препаратов, должен быть изложен в специальной инструкции. 30. Требования к исходным и упаковочным материалам, этикеткам и другим средствам маркировки, критическим промежуточным материалам и готовой продукции должны быть указаны в спецификациях. Спецификации также разрабатываются на критические материалы и компоненты (вспомогательные вещества, уплотнения, наборы для стерилизующей фильтрации и др.), используемые в процессе и способные оказать критическое влияние на качество продукции. 31. Следует установить допустимые предельные значения изменений характеристик радиофармацевтических препаратов, включая требования к выпуску и сроку хранения (например, радиохимической чистоты, объемной активности, радионуклидной чистоты и удельной активности). 32. В протоколах на использование, очистку, дезактивацию, дезинфекцию (стерилизацию), техническое обслуживание следует указывать дату и время выполнения операции, заверять протокол подписью лица, выполнившего работу, и, при необходимости, указывать наименование продукта и номер серии. 33. Протоколы следует хранить не менее чем в течение трех лет, если иное не указано в других документах.
Производство
34. Одновременное производство различных радиофармацевтических препаратов в одной рабочей зоне (горячей камере, ламинарной зоне или шкафу) не допускается, что вызвано необходимостью снижения до минимума риска перекрестного загрязнения радиоактивными веществами или перепутывания материалов. 35. Процессы и оборудование подлежат аттестации (испытаниям), в т.ч. системы с компьютерным управлением, в соответствии с Приложением 11 к настоящему стандарту. Новые процессы подлежат перспективной аттестации. 36. Критические параметры следует, как правило, определить до проведения аттестации или в ее процессе. При этом следует определить допустимые предельные значения изменений параметров, необходимые для стабильного производства. 37. Для продуктов, наполняемых в асептических условиях, следует проводить контроль целостности мембранных фильтров, принимая во внимание необходимость обеспечения радиационной безопасности и сохранения стерильности фильтров. 38. Учитывая радиационную активность готового продукта, допускается наносить маркировку на первичную упаковку до начала производства. На стерильные пустые закрытые флаконы может быть нанесена маркировка с частичной информацией до операции наполнения, при этом стерильность не должна быть нарушена и не должно быть помех для визуального контроля наполненных флаконов.
Контроль качества
39. Некоторые радиофармацевтические препараты могут быть выпущены и использованы на основе оценки документации на серию до завершения всех химических и микробиологических испытаний. Оформление разрешения на выпуск радиофармацевтических препаратов может быть выполнено в два и более этапов до и после завершения аналитического контроля в полном объеме: a) разрешение на выпуск оформляют после оценки протокола на серию, в котором должны быть отражены условия производства и результаты аналитического контроля; препарат до отправки в клинику имеет статус "в карантине"; b) получение разрешения на выпуск от уполномоченного лица после проведения оценки окончательных результатов аналитического контроля, всех отклонений от нормального процесса, которые должны быть оформлены документально, обоснованы и утверждены. Если некоторые результаты контроля невозможно получить до использования препарата, то уполномоченному лицу следует оформить разрешение на выпуск препарата условно до его использования и окончательно оформить разрешение на выпуск препарата после получения всех результатов контроля. 40. Большинство радиофармацевтических препаратов используются в течение короткого периода времени. Срок годности препарата должен быть четко указан. 41. Препараты, содержащие радиоактивные изотопы с большим периодом полураспада, следует контролировать на соответствие всем требованиям до оформления разрешения на выпуск уполномоченным лицом. 42. Контроль проб может быть проведен не сразу после их отбора, чтобы обеспечить требуемое снижение уровня активности. Все виды контроля, включая контроль на стерильность, должны быть проведены как можно быстрее. 43. Порядок оценки продукции и результатов контроля, которую следует провести до выпуска продукции, должен быть изложен в специальной инструкции. 44. Продукция, не соответствующая установленным требованиям, должна быть отклонена. Если предусмотрена переработка материала, то она должна выполняться по инструкции. Готовая продукция должна соответствовать установленным требованиям, что должно быть подтверждено до ее выпуска. Не допускается переработка возвращенной продукции, с которой следует обращаться как с радиоактивными отходами. 45. В специальной инструкции должен быть определен порядок действий уполномоченного лица в случае обнаружения несоответствия продукции требованиям спецификации после ее отгрузки до истечения срока годности. Такие случаи должны быть расследованы с разработкой плана мероприятий по их предупреждению и документальным оформлением. 46. При необходимости следует информировать ответственный персонал лечебного учреждения. Это следует выполнять в порядке, обеспечивающем прослеживаемость всех действий. 47. Должен быть разработан порядок контроля исходных материалов. При выборе и утверждении поставщика следует убедиться в том, что поставляемые им материалы неизменно соответствуют требованиям спецификаций. Исходные и упаковочные материалы и вспомогательные материалы для критических процессов должны приобретаться только у утвержденных поставщиков.
 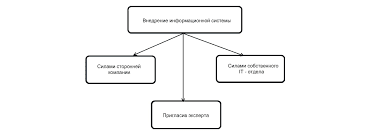 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)... 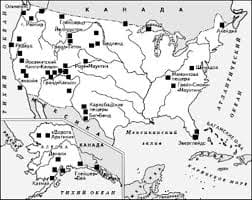 Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования...  Конфликты в семейной жизни. Как это изменить? Редкий брак и взаимоотношения существуют без конфликтов и напряженности. Через это проходят все... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|