
|
|
НОРМАТИВНО-ТЕХНІЧНА ДОКУМЕНТАЦІЯ У ПРОМИСЛОВОМУ ВИРОБНИЦТВІ ЛІКІВПромислове виробництво ліків регламентується відповідною нормативно-технічною документацією (НТД), затвердженою за встановленим порядком. НТД має забезпечувати підвищення якості та ефективності лікарських препаратів, постійно удосконалюватися на основі досягнень науки і техніки і вчасно переглядатися з метою заміни застарілих показників відповідно до потреб охорони здоров'я населення, оборони країни та експорту. В Україні існують однакові вимоги до змісту, порядку розробки, погодження і затвердження НТД на хіміко-фармацевтичну продукцію медичного призначення, а також продукцію ветеринарного призначення і харчові добавки, вироблені хіміко-фармацев-тичними підприємствами й фармацевтичними фабриками. Нормативна документація — це документи, що встановлюють правила, загальні принципи або характеристики, що стосуються різних видів діяльності або її результатів. НТД на лікарські препарати, лікарську рослинну сировину і вироби медичної техніки поділяють на такі категорії: 1. Технологічні і технічні регламенти. 2. Державна фармакопея (ДФ). 3. Аналітична нормативна документація. 4. Державні стандарти (ГОСТ, ДСТ У).
5. Галузеві стандарти (ОСТ), Галузевий стандарт України (ГСТ У). 6. Технічні умови (ТУ У). 7. Керівний нормативний документ (КД) — інструкції, методичні вказівки тощо. 8. Виробничі технологічні інструкції. Аналітична нормативна документація (АНД) — фармакопейні статті, документи про методи аналізу, а також інша аналітична документація, яка дозволяє контролювати якість лікарського засобу. АНД є невід'ємною частиною реєстраційних документів — комплекту матеріалів на лікарський засіб, спеціалізована оцінка яких надає змогу зробити висновки про можливість його державної реєстрації, потребу проведення передреєстраційних досліджень або контролю якості зразків лікарського засобу. Проведення експертизи та затвердження АНД регламентує Постанова Кабінету Міністрів України від 13 вересня 2000 року за № 1422 «Порядок державної реєстрації (перереєстрації) лікарського засобу». АНД повинна містити такі відомості: — склад препарату із зазначенням точної кількості усіх інґре-дієнтів на одиницю лікарського засобу з посиланням на монографії з фармакопей, яким вони відповідають за якістю. Якщо діючу або допоміжну речовину не описано в фармакопеях, то потрібно надавати на них інші категорії АНД (ДСТ У, ТУ У і т. д.); — для органопрепаратів, отриманих за допомогою генної інженерії або іншим оригінальним способом, необхідно вказати назву сировини, з якої виготовляють препарат, спосіб його одержання, а також АНД на сировину; — специфікацію у вигляді таблиці, в якій у першій колонці перераховані всі показники якості препарату, у другій — наведена регламентація за цими показниками, а в третій колонці вказані посилання на методи контролю за цими показниками; — методики контролю якості препарату фірми-виробника за порядком, наведеним в специфікації; — упаковку, маркування, транспортування, зберігання, термін придатності. В кінці АНД наводять відомості про основну фармакологічну дію лікарського засобу. Затверджується АНД Наказом Міністерства охорони здоров'я України зі зазначенням номера реєстраційного посвідчення лікарського засобу і підписується директором Фармакологічного центру МОЗ України. На підставі рішення про державну реєстрацію лікарський засіб вноситься до Державного реєстру лікарських засобів, що ведеться Міністерством охорони здоров'я України. Матеріали щодо методів контролю (АНД) за якістю лікарського засобу надсилаються до Державного департаменту з контролю за якістю, безпекою та виробництвом лікарських засобів і виробів медичногопризначення та Державної інспекції з контролю за якістю лікарських засобів. Чинність АНД (в часі) визначається терміном дії реєстрацій Стандарт — нормативний документ, в якому встановлено для загального і багаторазового використання правила, вимоги, загальні принципи або характеристики, що стосуються різних видів діяльності або їх результатів для досягнення оптимального ступеня упорядкованості в зазначеній галузі. Державний і галузевий стандарти (ДСТ У, ГСТ У) установлюють на додаткові технічні вимоги та групові характеристики, необхідні для виготовлення і постачання лікарських препаратів (технічні терміни і позначення, загальнотехнічна документація, технологічні норми тощо). Державні стандарти затверджуються Міністерством охорони здоров'я України або Міністерством медичної і мікробіологічної промисловості України за погодженням із МОЗ України. Деякі види сировини, допоміжні речовини, тара та упаковка нормуються технічними умовами (ТУ У). Подібно до статей у фармакопеї ТУ У мають характер державного стандарту. Технічні умови — нормативний документ, що встановлює вимоги до конкретної продукції чи послуг і регулює відносини між постачальником та споживачем продукції. Уся робота фармацевтичних підприємств відзначається суворою регламентацією і плануванням виробництва. Технологічний процес виробництва лікарських препаратів здійснюється на підставі нормативно-технічної документації, наданої у вигляді двох регламентів: технологічного, що має відношення до виробництва конкретного найменування продукції, та технічного, що містить вимоги до комплексу обладнання і його безпечної експлуатації на певній виробничій ділянці певного цеху. Вимоги цих регламентів гарантують якість продукції, що випускається, раціональне й безпечне здійснення технічних проце-
сів, збереження обладнання, виключення причин виникнення аварій і забруднення навколишнього середовища. Технологічний регламент — це нормативний документ, в якому викладено технологічні методи, технічні засоби, норми і нормативи виготовлення лікарського засобу. Технічний регламент — це нормативний документ, в якому для конкретного комплексу технологічного устаткування викладено умови, що забезпечують випуск напівпродуктів або лікарських засобів окремої лікарської форми заданої якості. Технологічний регламент поширюється на виробництво конкретного лікарського препарату в умовах, продиктованих технічним регламентом. Дія технічного регламенту охоплює підготовку виробничих (лабораторних,дослідно-промислових та промислових) приміщень і персоналу до роботи; створення необхідних санітарно-ґігієніч-них умов виробництва; виконання вимог, пов'язаних з охороною праці, технікою безпеки, пожежною безпекою, охороною навколишнього середовища; кваліфіковану ефективну експлуатацію устаткування, що гарантує.одержання лікарських засобів відповідно до Ъимог НТД. Регламент виробництва хіміко-фармацевтичної продукції використовують як основний технологічний документ: — при підготовці розроблюваної хіміко-фармацевтичної продукції для доклінічного і клінічного вивчення й постановки нової продукції на виробництво; — серійному виробництві хіміко-фармацевтичної продукції та напівпродуктів для неї; — складанні виробничих інструкцій з техніки безпеки, промислової санітарії та протипожежних заходів; — розробці та здійсненні заходів утилізації відходів виробництва, знешкодження та очищення промислових стоків та викидів в атмосферу; — встановленні техніко-економічних нормативів, у тому числі норм витрачання сировини та матеріалів; — проектуванні промислового виробництва. Залежно від стадії розробки продукції, ступеня освоєння технології виробництва або мети здійснюваних робіт регламенти бувають двох категорій: — технологічні тимчасові регламенти (TTP); — технологічні промислові регламенти (ТПР). За тимчасовими технологічними регламентами виконують лабораторні й дослідно-промислові роботи, виготовляють пробні партії лікарських засобів для проведення доклінічних і клінічних досліджень. TTP є документом на право одержання дозвопу до медичного застосування лікарських препаратів і затвердження тимчасової фармакопейної статті. За TTP дозволяється реєструвати і робити разові та промислові серії лікарських препаратів для оптової реалізації при невеликих обсягах виготовлення продукції, що встановлюються окремим рішенням Технологічної комісії Держкоммедбіопрому. Термін дії TTP — до трьох років. У відповідності з технологічними промисловими регламентами здійснюється серійне виробництво хіміко-фармацевтичної продукції; ТПР є основним документом для реєстрації лікарського препарату в Україні. Термін дії ТПР — не більше п'яти років. Технологічний регламент незалежно від типу має містити розділи, а саме: 1. Характеристика готової продукції. 2. Схеми виробництва і технологічного процесу:
— блок-схема виробництва; — характеристика сировини, матеріалів і напівфабрикатів; — опис стадій технологічного процесу; ч — матеріальний баланс.
3. Контроль виробництва. 4. Додатки:
— перелік технологічних інструкцій виготовлення; — перелік форм протоколів. Технічний регламент має такі розділи:
1. Загальна характеристика виробництва. 2. Апаратурна схема, специфікація устаткування і контрольно-вимірювальних приладів. 3. Експлуатація технологічного обладнання і контрольно-вимірювальних приладів. 4. Загальна схема системи контролю якості. 5. Безпечна експлуатація виробництва та охорона навколишнього середовища. 6. Загальний перелік виробничих інструкцій. 7. Інформаційні матеріали:
— додаток про технічний стан виробництва; — інформаційний додаток про лікарський засіб; — протоколи валідації виробництва. Дотримання всіх вимог технологічного регламенту обов'язкове. Регламент є законом виробництва і відступати від нього неприпустимо. МАТЕРІАЛЬНИЙ БАЛАНС При виробництві готових лікарських засобів кількість готового продукту разом із побічними продуктами й відходами завжди менша за кількість вихідних матеріалів із-за наявних на кожному виробництві матеріальних втрат, рівень яких залежить від досконалості технологічного процесу.
Це явище ілюструється так званим рівнянням матеріального балансу, що має вигляд: Сі=(С2+Сз+С4) + С5. Матеріальний баланс — співвідношення між кількістю вихідної сировини, матеріалів, напівпродуктів і проміжної продукції (С1), використаних у виробництві, і кількістю фактично отриманої готової продукції (С2), побічних продуктів (С3), відходів або покидьків (С4) і втрат (C-), тобто співвідношення теоретично можливого і практично отриманого виходу готової продукції. У разі, якщо побічні продукти виробництва відсутні, рівняння матеріального балансу спрощується: Сх = С2 + С5. Матеріальні втрати при виробництві лікарських препаратів бувають різного походження, тому їх розподіляють на кілька груп: — механічні, які виникають здебільшого при відсутності або недостатній механізації переміщення матеріалів під час переробки (проливання рідини, розпилення, утруска, бій і т. ін.); — фізико-хімічні, що спостерігаються у разі проведення технологічного процесу без урахування фізико-хімічних властивостей лікарських речовин (неповне екстрагування діючих речовин із лікарської рослинної сировини, втрати легколетких розчинників під час фільтрації, ефірного масла при випарюванні тощо); — хімічні, що можливі внаслідок недотримання або неправильного вибору параметрів проведення хімічних реакцій (синтезу). Матеріальний баланс має велике практичне значення, тому що він зумовлює ступінь досконалості технологічного процесу. Чим повніше він складений, тим детальніше вивченою є технологія даного препарату. Чим менше в балансі різного роду втрат, тим правильніше здійснюється процес виробництва. I навпаки — чим більше в балансі матеріальних втрат, тим менш досконалою вважається технологія цього препарату. Матеріальний баланс лежить в основі регламенту виробництва і дає можливість оцінити рівень організації технологічного процесу, порівняти ефективність його проведення на різних виробництвах, що випускають однойменну продукцію. Матеріальний баланс складається на підставі проведеного експерименту на одиницю продукції, що випускається, на один виробничий потік або потужність усього виробництва. Для нових виробництв його складають за даними проекту, для діючих — за досягнутими показниками роботи в останній рік перед затвердженням регламенту. Його переглядають тільки в разі включення у технологічний процес (чи виключення з нього) операцій або стадій, що значно впливають на витрати сировини чи кількість відходів. Матеріальний баланс може бути поданий як у вигляді алгебраїчного рівняння, так і у формі таблиць надходження й витрат матеріалів, що є характерним для технологічних регламентів виробництва готових лікарських засобів. У прибутковій частині балансу зазначається кількість матеріалів, уведених у виробництво, а у видатковій частині — кількість отриманих матеріалів і втрат. У підсумку прибуткова і видаткова частини балансу повинні складати однакові суми. Складається матеріальний баланс як для технологічного процесу загалом, так і на кожну окрему стадію або технологічну операцію (постадійний матеріальний баланс). Він може охоплювати всі матеріали (загальний, сумарний баланс) або кожний окремий компонент. Матеріальний баланс складається на одну серію продукції за об'єктивними результатами запропонованого рівня технології виготовлення лікарських засобів. У регламентах виробництва слід подавати дві таблиці матеріального балансу для кожної серії продукції. У першій таблиці для кожної стадії технологічного процесу наводяться дані про види й кількість (масу, об'єм, молярну масу, штуки) сировини, напівпродуктів, матеріалів, відходів і готових продуктів, витрачених і отриманих у процесі виробництва. Послідовність стадій виробництв відображається блок-схемою технологічного процесу. Таблиця складається для однієї серії готової продукції (напівпродукту). Визначення серії подано у відповідній нормативній документації. Постадійний матеріальний баланс можна відобразити у вигляді таблиці 2.1 (приклад виробництва настойки звіробою). Залежно від особливостей сировини баланс на деякі стадії виробництва ведуть не тільки за технічною масою матеріалів (графа 3), але й за кількістю основної речовини (графа 4). Наприклад, для спирту — за абсолютним спиртом, для таблеток, капсул, мазей і т. д. — за вмістом діючих речовин, для рослинної сировини — за екстрактивними речовинами, вологою або діючими речовинами. Наводимо приклад розрахунку спирту 96 % -вого для одержання настойки. Розрахунок здійснюють таким чином: Технічна маса 96 % -вого спирту етилового на одну серію складає 389,48 кг. Об'єм 96 %-вого спирту етилового при цьому складатиме 482,33 л (V= m/p, 389,48/0,8075). Знаходимо кількість абсолютного спирту в літрах, що міститься в 389,48 кг 96 %-вого спирту. В алкогометричній таблиці № 1 ДФ XI (вип. 1) знаходимо, що в 100 г 96 %-вого спирту міститься 119,04 мл абсолютного спирту, отже, в 389,48 кг міститься 463,63 л абсолютного спирту.
Таблиця 2.1 Виробництво настойки звіробою До таблиці записуємо отримані результати:
Об'єми розчинів різних концентрацій (графа 6) зазвичай не підсумовують у зв'язку з можливим явищем контракції. Під найменуваннями відходів і втрат указують сировину, матеріали і напівпродукти, що містяться в них. За наявності у відходах і втратах великої кількості інґредієнтів дозволяється не перелічувати їх, а лише вказати сумарну кількість. Графа 5 заповнюється для виробництв, пов'язаних із хімічними перетвореннями, а графа 7 — для виробництв готових лікарських засобів. Значення об'єму рідин у графі 6 не виключає даних про їх маси в графах 3—5. Матеріальний баланс складається з припустимих показників якості сировини, а при завантаженні здійснюється перерахунок на їх вміст у відповідності з даними аналізу. Фактичні дані відображаються в операційному листку (протоколі виготовлення). Зазначені в матеріальному балансі втрати — це межа, яку не можна перевищувати. Зведений матеріальний баланс серії продукції подається також у вигляді таблиць (табл. 2.2), в яких наводяться відомості про види й кількість витраченої сировини та напівпродуктів (колонка «Витрачено») і отриманих продуктів у процесі роботи (колонка «Одержано»). При складанні таблиці можливі варіанти з використанням одиниць вимірювання, введенням додаткових колонок (кількість у штуках, маса в кілограмах тощо). У графах 3 і 4 після найменування кінцевого продукту і зазначення його кількості вказують найменування і сумарний вміст відходів. Після кожної із цих позицій подають найменування і кількість речовин, які містяться відповідно у відходах. За матеріальним балансом розраховують основні техніко-еко-номічні показники виробництва — такі як регламентовані норми витрат сировини, матеріалів, напівпродуктів та енергоресурсів на одиницю продукції. Розрахунок норм витрат сировини виконують за фактичними даними необхідного статистичного набору експериментів, з урахуванням використаного обладнання, неминучих виробничих відходів і втрат при суворому дотриманні технологічного процесу, скориставшись формулою: тт Кількість завантаженої речовини Норма витрат =------------------------------------------- ---------------. Кількість отриманої речовини Таблиця 2.2 Матеріальний баланс серії настойки звіробою для фасування у флакони по 100 мл
Якщо регламентом передбачається повернення сировини і регенерація, норма витрат розраховується таким чином: тт (К-сть завант. реч. - К-сть поверненої вих. реч.) (К-сть отрим. реч. - К-сть регенерованої реч.) Норми витрат сировини, матеріалів, напівпродуктів та енергоресурсів розраховуються з точністю до 0,001. Наводимо приклад розрахунку норми витрат спирту етилового при одержанні настойки зверобою: На стадії ТП2. Одержання настойки витрачено (за абсолютним спиртом): — 13,710 л абсолютного спирту з відгону; — 463,630 л абсолютного спирту з водно-спиртової суміші. Отримано на стадії ТП2: — 452,840 л абсолютного спирту в настойці; — 14,910 л абсолютного спирту в шроті; — 9,590 л абсолютного спирту у втратах. Запишемо рівняння матеріального балансу: 13,710 + 463,630 = 452,840 + 14,910 + 9,590. Оскільки шрот підлягає регенерації, то на стадії переробки відходів із 14,91 л абсолютного спирту одержимо 13,71 л абсолют- ного спирту з відгону. Рівняння матеріального балансу матиме такий вигляд: 14,910= 13,710+ 1,200. Отже, норма витрат абсолютного спирту на стадії ТП2. Одержання настойки складатиме: 477,340 Норма витрат =--------------------------------- = 1,023. 452,840 + 13,710 При складанні матеріального балансу можуть бути наведені інші показники й нормативи, що визначають технічний рівень та ефективність виробництва, наприклад, регламентний видатковий коефіцієнт, ступінь використання сировини, зняття продукції з одиниці виробничої площі або з одиниці вартості основних фондів та інші показники. ОСНОВНІ ПОЛОЖЕННЯ GMP Третя ланка системи забезпечення якості пов'язана з гарантуванням якості лікарських препаратів за рахунок виробництва у відповідності з правилами GMP. Ця ланка ще не дуже розвинута. Закон України «Про лікарські засоби» (ст. 11) передбачає виробництво лікарських засобів з урахуванням міжнародних норм. За ДСТУ 1.0—93 одним з основних завдань міжнародного науково-технічного співробітництва України в галузі стандартизації є зближення і гармонізація української державної системи стандартизації з міжнародними і регіональними системами, а також із прогресивними національними системами стандартизації інших країн. 3 огляду на це виникла нагальна потреба в розробці документа «Виробництво лікарських засобів. Належні правила і контроль якості», який мав би відповідати принципам і вимогам GMP ВООЗ і ЄС, а також враховувати законодавство і умови нашої країни. Тепер в Україні прийнято до виконання варіант GMP ЄС, в якому викладено принципи і правила виробництва й контролю якості лікарських препаратів. У 1991 році Комісією ЄС були прийняті дві директиви, які містять принципи і вказівки щодо належного виробництва лікарських препаратів, призначених для застосування в медицині (директива 91/356/ЄЕС) і у ветеринарії (директива 91/412/ЄЕС). У цих директивах належна виробнича практика (GMP) була ратифікована як невід'ємна частина національних систем забезпечення якості лікарських препаратів у країнах — членах ЄС.
Директивами встановлено основні принципи GMP щодо виробництва лікарських засобів, а саме таких його сторін: 1. Управління якістю. 2. Персоналу. 3. Приміщень та обладнання. 4. Документації. 5. Виробництва. 6. Контролю якості. 7. Робіт за контрактом. 8. Рекламацій та відкликання продукції. 9. Самоінспекції. Слід зазначити, що принципи і правила GMP ЄС і ВООЗ практично ідентичні. Документи відрізняються тільки порядком викладу принципів і правил. GMP ЄС складається з таких розділів: — Вступ. — Галузь застосування. — Нормативні посилання. — Визначення.. — Позначення і скорочення. — Вимоги до управління якістю. — Вимоги до персоналу. — Вимоги до приміщень та обладнання. — Вимоги до документації. — Вимоги до виробництва. — Вимоги до контролю якості. — Вимоги до виробництва та іспитів, що виконуються за контрактом. — Рекламації та відкликання продукції. — Вимоги до самоінспекції. Крім того, у цьому документі є додатки: Додаток 1. Виробництво стерильної медичної продукції. Додаток 2. Виробництво біологічної медичної продукції для людей. Додаток 3. Виробництво радіоактивних фармацевтичних препаратів. Додаток 4. Виробництво ветеринарної медичної продукції, крім імунологічної ветеринарної медичної продукції. Додаток 5. Виробництво імунологічної ветеринарної медичної продукції. Додаток 6. Виробництво медичних газів. Додаток 7. Виробництво рослинної медичної продукції. Додаток 8. Відбір проб вихідних і пакувальних матеріалів. Додаток 9. Виробництво рідин, кремів і мазей. Додаток 10. Виробництво аерозолей для інгаляцій. Додаток 11. Комп'ютерні системи. Додаток 12. Застосування іонізуючого випромінювання у виробництві медичної продукції. Додаток 13. Практика якісного виробництва медичної продукції для клінічних випробувань. Додаток 14. Виробництво продукції з крові або плазми людини. Основні принципи GMP. Головний принцип GMP полягає в тому, що виробник лікарських засобів повинен створити і впровадити ефективну систему забезпечення якості, включаючи активну участь дирекції та всього персоналу. Система якості — це сукупність організаційної структури, методик, процесів і ресурсів, необхідних для здійснення процесу управління якістю. Стандарт GMP призначений для побудови систем якості на підприємствах, які виробляють лікарські засоби. У розділі 1 «Управління якістю» викладено фундаментальну концепцію системи забезпечення якості під час виробництва лікарських засобів. У наступних розділах її принципи і правила розглядаються детальніше, щоб їх можна було адекватно трактувати, а також успішно застосовувати при розробці і впровадженні на підприємствах-виробниках систем якості. Основний принцип стосовно персоналу такий: оскільки система якості і виробництво залежать від людей, то штат має бути укомплектований достатньою кількістю кваліфікованого персоналу, який здатний на належному рівні вирішувати всі завдання, що знаходяться у сфері відповідальності підприємства. Кожний співробітник повинен чітко знати свої повноваження і обов'язки, а також усвідомлювати індивідуальну відповідальність (їх слід відображати в посадових інструкціях), знати і суворо дотримувати правил GMP при виконанні посадових обов'язків. Усі співробітники при вступі на посаду зобов'язані пройти докладний інструктаж про принципи і правила GMP, включаючи правила особистої гігієни; потім у процесі діяльності вони мають регулярно підвищувати кваліфікацію. Наступний принцип стосується приміщень і обладнання, які необхідно проектувати, розміщувати, конструювати, оснащувати, пристосовувати, а також утримувати й обслуговувати таким чином, щоб вони відповідали своєму призначенню і були придатні для передбачених робіт. їхні розмір, конструкція і розташування мають зводити до мінімуму ризик помилок на виробництві і забезпечувати ефективне прибирання та експлуатацію з метою уникнення перехресної контамінації, накопичення пилу та інших забруднень, що можуть негативно вплинути на якість продукції. Якщо розташування приміщень і технічний рівень обладнання не забезпечують якість продукції, то потрібна їх модифікація.
Важливою частиною системи забезпечення якості є документація. Вона має регламентувати всі аспекти виробництва і контролю якості лікарських препаратів. Виробництво лікарських засобів повинне здійснюватися за технологічним регламентом, з урахуванням принципів і правил належної виробничої практики (GMP). Це необхідно для одержання готової продукції потрібної якості, що відповідала б вимогам реєстраційної та ліцензійної документації. Відповідність реєстраційної та ліцензійної документації і санкціонування істотних змін уповноваженими державними органами є найважливішим положенням усіх стандартів GMP і директив ЄС. Необхідними ланками виробництва є виробничий контроль і валідація. Валідація — це експертна оцінка та надання документально оформлених об'єктивних доказів у відповідності з принципами GMP, які підтверджують, що будь-які об'єкти дійсно відповідають своєму призначенню і встановленим вимогам, а їх використання веде до очікуваних результатів. Наступний принцип GMP належить до контролю якості. Контроль якості включає роботи, пов'язані з відбором проб, нормативною документацією (специфікаціями) та випробуваннями, а також із методиками організації цих робіт, їх документуванням і видачею у встановленому порядку дозволів, які гарантують, що всі необхідні випробування дійсно проведено. Вихідна сировина, матеріали, напівпродукти і проміжна продукція не дозволяються для використання, а готова продукція не допускається до реалізації доти, доки їх якість не буде визнана задовільною. Основною вимогою до контролю якості є його незалежність від виробництва. Окремий розділ GMP присвячений роботам, які виконуються за контрактом. У ньому йдеться про те, що при аналізі контракту всі умови виробництва і/або випробувань повинні бути чітко і всебічно визначені, узгоджені і проконтрольовані, щоб уникнути непорозумінь і невідповідностей, які можуть стати причиною незадовільної якості продукції, виконуваних робіт або випробувань. Важлива також наявність письмового контракту (договору), оформленого за встановленим порядком між двома юридичними особами, що іменуються відповідно Замовником і Виконавцем. Договір повинен мати юридичну силу, і в ньому треба чітко визначати права та обов'язки кожної із сторін, зокрема дотримання правил GMP. У контракті необхідно визначати й порядок видачі уповноваженою особою дозволу на реалізацію кожної серії продукції або сертифіката якості. Правила GMP розмежовують відповідальність між Виконавцем і Замовником перед уповноваженими державними органами, що здійснюють реєстрацію і ліцензування, але вони не стосуються обопільної відповідальності Замовника і Виконавця за якість продукції (послуг) перед споживачем, яку вони несуть згідно із законодавством України. Наступний принцип проголошує, що всі рекламації та інша інформація про невідповідність якості потенційно бракованої продукції повинні ретельно перевірятися за стандартною робочою методикою. На підприємстві-виробнику має бути організована система, що дозволяє в разі необхідності швидко та ефективно відкликати реалізовану продукцію, якщо в ній встановлені або можливі дефекти якості. I, нарешті, останній непорушний принцип: на підприємстві повинні діяти самоінспекція та аудит якості, призначення яких полягає у всебічному нагляді за виконанням правил GMP і, якщо необхідно, укладанні рекомендацій щодо проведення запобіжних та коригувальних дій. Якщо узагальнити правила GMP як єдиного документа, що регламентує систему якості підприємства, то суть їх така. Кожне окреме правило GMP цілком зрозуміле, але виконувати їх треба всі в комплексі, створюючи систему якості. Саме через порушення цього принципу не вдалося запровадити РД 64-125—91, що був позбавлений низки правил GMP, і тому припускав існування на підприємствах окремих елементів GMP, а не сучасних систем якості. Друга особливість полягає в тому, що правила GMP висувають вимоги, але не дають конкретних технічних рішень. Яскравим прикладом є вимоги до приміщень і обладнання. Наприклад: «Приміщення мають бути розташовані таким чином, щоб звести до мінімуму ризик контамінації» або: «Устаткування має відповідати своєму призначенню і передбаченому технологічному процесові». Технічне рішення залишається за підприємством, тобто керівництву і всьому колективу треба не просто виконувати «волю» стандарту, а виявляти творчий підхід, оскільки в стандартах GMP регламентовано, що саме потрібно зробити, але не зазначено, яким чином. Часто засоби реалізації технічних рішень виявляються дуже складними і дорогими. Складність зростає ще й тому, що ці засоби не повинні суперечити законодавству України, а також правовим нормативним актам. У зв'язку з цим виникла необхідність СНіПи 80-х років привести у відповідність із сучасним рівнем техніки. Тому з 01.01.1997 року проектування та будівництво нових, розширення діючих підприємств і виробничих об'єктів почали здійснювати тільки у відповідності з правилами GMP. Реконструкцію і технічне переоснащення підприємств з урахуванням правил GMP запроваджено з 01.06.1998 року. 3 01.01.2002 року правила GMP стають в Україні обов'язковими. Перехід на виробництво лікарських засобів за новими принципами і правилами здійснюватиметься поетапно, за графіками, які будуть розроблені для кожного підприємства.
3.1. ХАРАКТЕРИСТИКА I КЛАСИФІКАЦІЯ РОЗЧИНІВ Розчини — це рідкі гомогенні системи, які складаються із розчинника та одного або декількох компонентів, розподілених у ньому у вигляді іонів або молекул. Медичні розчини різноманітні за властивостями, складом, способами одержання і призначенням. Виготовляються вони переважно на фармацевтичних виробництвах системи аптечних управлінь Міністерства охорони здоров'я України. Окремі розчини, виготовлення яких вимагає проведення хімічних реакцій, одержують на хіміко-фармацевтичних заводах Міністерства медичної й мікробіологічної промисловості України (наприклад, рідина Бурова та ін.). Розчини мають багато переваг перед іншими лікарськими формами, тому що значно швидше всмоктуються у шлунково-кишковому тракті. А вадами розчинів є їх великий об'єм, можливі гідролітичні і мікробіологічні процеси, що спричиняють швидке руйнування готового продукту. Знання технології розчинів важливе при виготовленні майже всіх інших лікарських форм, де розчини є напівпродуктами або допоміжними компонентами. Розчини займають проміжне становище між хімічними сполуками і механічними сумішами. Від хімічних сполук розчини відрізняються змінністю складу, а від механічних сумішей — однорідністю. Тому розчинами називають однофазні системи змінного складу, утворені не менш як двома незалежними компонентами. Найважливіша особливість процесу розчинення — це його спонтанність. Достатньо простого зіткнення речовини з розчинником, щоб через деякий час утворилася однорідна система, тобто розчин. Розчинники можуть бути полярними і неполярними р<  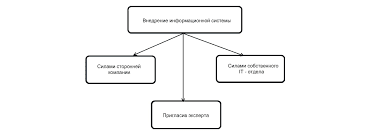 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала... 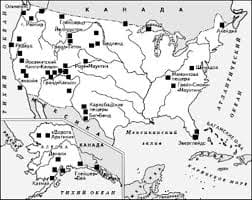 Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|
