
|
|
Основные теории катализа: мультиплетная, ансамблей, электронная, радикальная.Стр 1 из 3Следующая ⇒ Основные теории катализа: мультиплетная, ансамблей, электронная, радикальная. 1.Мультиплетная. Основные положения этой теории: 1.Активный центр кат-ра представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности в геометрическом соответствии со строением мол-лы, претерпевающей превращение(принцип геометрического или структурного соответствия). 2.При адсорбции реагирующих мол-л на активном центре образуется мультиплетный комплекс, в рез-те чего происходит перераспределение связей, приводящее к образованию продуктов реакции. Для различных реакций число адсорб.центров в активном центре принимается равным 2,3,4,6.подобные акт.центры были названы дублетами,триплетами,квадруплетами,секстетами, а в общем мультиплетами. Второе исх.положение явл. принцип энергетического соответсвия (Баландин),согласно к-ому энергия активации гетерог.р-ции явл.сложной вел-ной,в первом приближении состоящей из 2х слагаемых,одно из к-ых зависит только от энергии связи между составными частями реагир.в-в,а другое-от энергии вз-ия между катализатором и составными частями мультиплетного комплекса.Гетерог.р-ция представлена след.образом: Исх.в-ва+кат-тор→мультипл.комплекс→кат-тор+продукты р-ции Для I стадии необходима энергия разрыва связей,при этом выделяется энергии образования мультипл.комплекса.Е=разность этих энергий. Для II стадии необх.энергия для разрыва связей в мультипл.комплексе. выделяется энергия обр-ния конечных продуктов.Разность этих энергий определяет скорость II стадии. 2.Ансамблей Основное исх.положение – носителем каталит.активности явл. находящаяся на пов-ти атомная(докристаллич) фаза кат-ра,относительно к-ой пов-ть носителяч(или кристалл.фаза самого кат-ра) выполняет функцию инертной подкладки. Для каждого данного процесса акт.центром явл. ансамбль из определенного числа n атомов кат-ра. Теория акт.ансамблей дает возможность,исходя из опытной зав-ти активности от концентрации кат-ра на пов-ти носителя,определить число n атомов в акт.центре,число Zn, областей миграции и абсолют.производительность rn акт.центра для данного процесса. Зав-ть активности от концентрации
3.Электронная
65(2) мол-л реагента. Пр-с кат-за превращения включ.стадию хемосорбции на пов-ти тв.кат.,мол-ла реаг.в-ва- акцептор (дырка). Эл-ны или дырки уч.в созд.нов.мол-лы с пов-тью тв. тела. Акцепт.связь- образ.эл-ном пов-ти и акцепт.мол-лой (Сеα). Дон.связь- образ.своб. дыркой реш.тв.тела и донор.ат. Если ат.сорбир.без уч.эл-на или дырки-слабая связь. Зонная теор.строения тв.тел: при объедин.орбит.соседних ат.мет. возник.2 осн.зоны. Заполн.эн.ур-ни – валентная зона(система дырочн.ур-ней), вакант.эн.ур-ни- зона провод-сти(система эл.уровней. Эти зоны отделены запрещ. зоной. На пов-ти п/п может адс.акцепт.мол-ла, кот.притяг.эл-н из решетки,вал.зоны или на нее выпад.эл-н из зоны пров-сти.(Следовательно,происходит адсорбция на донорной или акцепторной связи е и дырки перемещ.поэтому слабые связи переходят в прочные и наоборот. Недостатки:хорошо для полупроводников,но не всегда применимо к др.в-вам.) 4.Радикальная На пов-ти мет.и п/п сод.радикалоподобные состояния ат.и мол. (нескомпенсир.вал-сти) Радикал(вал-сть) явл-ся нициат. цепи(обозн.V) Реакция.вида: A2+B2= 2AB Стадия а: A2+V à AV + A (инициир.цепи) Стадия б: A + V à (A) связыв.атома вак.вал-стью Стадия в: (A) + AV à A2 + V регенерация радикала Стадия г: B2+ V à BV + (B) Стадия д: (B) + BV à B2+ V Стадия е: (B) + V2à B +V ((В) может также реаг.на акт.ц. А): Стадия ж: (A) + BV à AB+V Стадия з: (B) +AV à AB +V; c А и В реаг.физич.адсорб.А2иВ2: Стадия и: (A) + (B2)à AB + (B) Стадия к: (B) +(A2) à AB + (A) Обрыв цепи-путем рекомб.своб.вал-стей: (A)+(A) = A2 (B)+(B) = B2 ((A)+(B) = AB V + V = V2 ск-сть отдельн.стадий р-ции завис.от велич.эн-гии связи A-V, B-V и теплот р-ций на стадиях а и г (Qav, Qbv). Теплоты вз-ия центров с мол-ми А2 и В2 опр-ся по разности между энергиями связей Qav и Qaa и Qbb q1=Qav-Qaa q`2=Qbv-Qbb Для кажд.пары эн-гии св. Qaa и Qbb пов-сть тв.кат.должна облад.меньшими велич. Е св. атомов с кат.ц. Qav и Qbv. 71. Влияние ионной силы на скорость реакции, проводимой в растворе. Во всех уравнениях химической кинетики фигурируют концентрации реагирующих веществ. В термодинамике же константа равновесия неидеальной системы выражается через активности. Это обстоятельство необходимо учитывать, если в кинетическое уравнение входит константа равновесия. Правда, при реакциях в газовой фазе и между нейтральными молекулами в растворе в этом нет практической необходимости, но при рассмотрении реакций между заряженными частицами подобное пренебрежение может явиться источником существенных ошибок. Для константы скорости бимолекулярной реакции A+ В → AВ* → продукты протекающей в растворе, теория активного комплекса, ;дает κ=χ Термодинамическая константа равновесия между исхешшми веществами и активным комплексом κ*= Комбинируя уравнения, получим уравнение Бренстеда — Бьеррума: κ=χ Если в качестве стандартного состояния выбрать бесконечное разведение, теория электролитов дает следующее приближенное соотношение для коэффициента активности: γ= Если zA и zB — соответствующие заряды реагирующих частиц, то реакция может быть записана в виде AZa+Bzb→AB*(Za+Zb) → продукты а подстановка соответствующих значений коэффициентов, активности в уравнение Бренстеда — Бьеррума дает Рассмотрим два случая. 1. Взаимодействие между заряженными частицами. Сумма Следовательно, логарифм константы скорости, согласно теории должен быть линейной функцией корня квадратного из ионной силы, а тангенс угла наклона прямой определяется соотношением зарядов реагирующих частиц. 2. Взаимодействие между ионом и нейтральной молекулой Согласно уравнению γ= получаем Следовательно, константа скорости должна быть прямо пропорциональна ионной силе. Зависимость константы скорости реакции от ионной силы называют первичным солевым эффектом. Вторичный солевой эффект (в случае слабых электролитов) – скорость реакции изменяется вследствие изменения одного из реагирующих присутствующих посторонних электронов, изменяющих ионную силу раствора и соответственно степень диссоциации реагирующего электролита.
72. Влияние природы растворителя и давления на скорость химической реакции, проводимой в растворе. При рассмотрении процессов в растворах необходимо учитывать структуру и природу растворителя и растворенного вещества, структуру, химический состав раствора и условия протекания реакции. Растворители бывают полярные или неполярные Растворы: ионные и молекулярные, концентрированные и разбавленные, идеальные и неидеальные. Структура чистых жидкостей – пространственные группы молекул (тетраэдр, тригональная бипирамидальная, октаэдр и др). Из сочетания 2-х, 3-х и более полиэдров создается ансамбль полиэдров. Число молекул растворителя, окружают такую же молекулу или ион – координационное число. При растворении в растворителе жидкости, газа или тв. вещества происходит их диспергирование до молекул или ионного состояния. Молекулы или ионы растворенного вещества занимают пустоты в полиэдрах или разрушая их, создают новые. Реагирующее вещество вступает не в форме индивидуальных молекул, а в форме сольватов. Молекула движется в «клетке» из молекул растворителя, движение колебательное и, следовательно, число столкновений больше, чем в газе, т. к. молекулы плотно упакованы в жидкости. W= kcini, k зависит от природы растворителя. Мономолекулярные реакции в жидкостях и в газовой фазе происходят с одинаковыми скоростями. Пусть в растворе протекает реакция: A+B По ЗДМ Концентрация переходного состояния:
Для мономолек.р-ции A ó A* -> продукты
Конст. равновесия
По теории АК:
Если график зависимости lnk от давления прямая,то ΔV не зависит от давления. Получим: Основные теории катализа: мультиплетная, ансамблей, электронная, радикальная. 1.Мультиплетная. Основные положения этой теории: 1.Активный центр кат-ра представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности в геометрическом соответствии со строением мол-лы, претерпевающей превращение(принцип геометрического или структурного соответствия). 2.При адсорбции реагирующих мол-л на активном центре образуется мультиплетный комплекс, в рез-те чего происходит перераспределение связей, приводящее к образованию продуктов реакции. Для различных реакций число адсорб.центров в активном центре принимается равным 2,3,4,6.подобные акт.центры были названы дублетами,триплетами,квадруплетами,секстетами, а в общем мультиплетами. Второе исх.положение явл. принцип энергетического соответсвия (Баландин),согласно к-ому энергия активации гетерог.р-ции явл.сложной вел-ной,в первом приближении состоящей из 2х слагаемых,одно из к-ых зависит только от энергии связи между составными частями реагир.в-в,а другое-от энергии вз-ия между катализатором и составными частями мультиплетного комплекса.Гетерог.р-ция представлена след.образом: Исх.в-ва+кат-тор→мультипл.комплекс→кат-тор+продукты р-ции Для I стадии необходима энергия разрыва связей,при этом выделяется энергии образования мультипл.комплекса.Е=разность этих энергий. Для II стадии необх.энергия для разрыва связей в мультипл.комплексе. выделяется энергия обр-ния конечных продуктов.Разность этих энергий определяет скорость II стадии. 2.Ансамблей Основное исх.положение – носителем каталит.активности явл. находящаяся на пов-ти атомная(докристаллич) фаза кат-ра,относительно к-ой пов-ть носителяч(или кристалл.фаза самого кат-ра) выполняет функцию инертной подкладки. Для каждого данного процесса акт.центром явл. ансамбль из определенного числа n атомов кат-ра. Теория акт.ансамблей дает возможность,исходя из опытной зав-ти активности от концентрации кат-ра на пов-ти носителя,определить число n атомов в акт.центре,число Zn, областей миграции и абсолют.производительность rn акт.центра для данного процесса. Зав-ть активности от концентрации
3.Электронная
65(2) мол-л реагента. Пр-с кат-за превращения включ.стадию хемосорбции на пов-ти тв.кат.,мол-ла реаг.в-ва- акцептор (дырка). Эл-ны или дырки уч.в созд.нов.мол-лы с пов-тью тв. тела. Акцепт.связь- образ.эл-ном пов-ти и акцепт.мол-лой (Сеα). Дон.связь- образ.своб. дыркой реш.тв.тела и донор.ат. Если ат.сорбир.без уч.эл-на или дырки-слабая связь. Зонная теор.строения тв.тел: при объедин.орбит.соседних ат.мет. возник.2 осн.зоны. Заполн.эн.ур-ни – валентная зона(система дырочн.ур-ней), вакант.эн.ур-ни- зона провод-сти(система эл.уровней. Эти зоны отделены запрещ. зоной. На пов-ти п/п может адс.акцепт.мол-ла, кот.притяг.эл-н из решетки,вал.зоны или на нее выпад.эл-н из зоны пров-сти.(Следовательно,происходит адсорбция на донорной или акцепторной связи е и дырки перемещ.поэтому слабые связи переходят в прочные и наоборот. Недостатки:хорошо для полупроводников,но не всегда применимо к др.в-вам.) 4.Радикальная На пов-ти мет.и п/п сод.радикалоподобные состояния ат.и мол. (нескомпенсир.вал-сти) Радикал(вал-сть) явл-ся нициат. цепи(обозн.V) Реакция.вида: A2+B2= 2AB Стадия а: A2+V à AV + A (инициир.цепи) Стадия б: A + V à (A) связыв.атома вак.вал-стью Стадия в: (A) + AV à A2 + V регенерация радикала Стадия г: B2+ V à BV + (B) Стадия д: (B) + BV à B2+ V Стадия е: (B) + V2à B +V ((В) может также реаг.на акт.ц. А): Стадия ж: (A) + BV à AB+V Стадия з: (B) +AV à AB +V; c А и В реаг.физич.адсорб.А2иВ2: Стадия и: (A) + (B2)à AB + (B) Стадия к: (B) +(A2) à AB + (A) Обрыв цепи-путем рекомб.своб.вал-стей: (A)+(A) = A2 (B)+(B) = B2 ((A)+(B) = AB V + V = V2 ск-сть отдельн.стадий р-ции завис.от велич.эн-гии связи A-V, B-V и теплот р-ций на стадиях а и г (Qav, Qbv). Теплоты вз-ия центров с мол-ми А2 и В2 опр-ся по разности между энергиями связей Qav и Qaa и Qbb q1=Qav-Qaa q`2=Qbv-Qbb Для кажд.пары эн-гии св. Qaa и Qbb пов-сть тв.кат.должна облад.меньшими велич. Е св. атомов с кат.ц. Qav и Qbv.   ЧТО ПРОИСХОДИТ, КОГДА МЫ ССОРИМСЯ Не понимая различий, существующих между мужчинами и женщинами, очень легко довести дело до ссоры...  Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|

 Для полупров-ков.Эл-ны-носит.эл-ва, при перемещ.в тв.теле остав.дырки(вакан.орбит.) Ток возможен если недостат.эл-ов в связях или в реш.есть изб.эл-нов. При электрич-ве эл-ны и дырки движ.в разл.направл. Они вызыв.превращ.акцепт.и дон.
Для полупров-ков.Эл-ны-носит.эл-ва, при перемещ.в тв.теле остав.дырки(вакан.орбит.) Ток возможен если недостат.эл-ов в связях или в реш.есть изб.эл-нов. При электрич-ве эл-ны и дырки движ.в разл.направл. Они вызыв.превращ.акцепт.и дон.
 =
= 
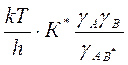 =К0
=К0 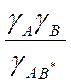 для константы скорости реакции, протекающей в любой неидеальной среде. Величина К0 имеет смысл константы скорости при бесконечном разведении, т. е. при γ= 1.
для константы скорости реакции, протекающей в любой неидеальной среде. Величина К0 имеет смысл константы скорости при бесконечном разведении, т. е. при γ= 1. ,где А — постоянная≈ 0,51; z —заряд иона; I— ионная сила раствора; β — некоторая постоянная, примерно обратно пропорциональная радиусу иона.
,где А — постоянная≈ 0,51; z —заряд иона; I— ионная сила раствора; β — некоторая постоянная, примерно обратно пропорциональная радиусу иона.
 мала, и для случая взаимодействия между двумя ионами ими можно пренебречь, т. е.
мала, и для случая взаимодействия между двумя ионами ими можно пренебречь, т. е. 
 , коэффициент активности для нейтральной молекулы γ=
, коэффициент активности для нейтральной молекулы γ=  и для реакции A+Bzb→AB*(Zb) → продукты
и для реакции A+Bzb→AB*(Zb) → продукты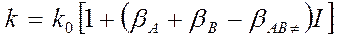
 A..B
A..B 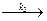 C, раствор неидеальный.
C, раствор неидеальный.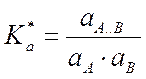 или
или 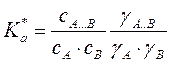 ,где a и y – активности и коэффициенты активности
,где a и y – активности и коэффициенты активности . По теории переходного состояния
. По теории переходного состояния  , h-постоянная Планка При
, h-постоянная Планка При  получим
получим 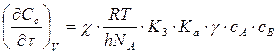 Скорость химической реакции зависит от природы растворителя. Так при y=10 скорость реакции в растворе в 10 раз выше чем в газовой фазе.При y=1 скорость реакции в растворе = скорости в газовой фазе.
Скорость химической реакции зависит от природы растворителя. Так при y=10 скорость реакции в растворе в 10 раз выше чем в газовой фазе.При y=1 скорость реакции в растворе = скорости в газовой фазе. и при
и при 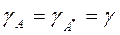 получим:
получим: , т.е. скорость реакции не зависит от природы растворителя.
, т.е. скорость реакции не зависит от природы растворителя. (V-изменяется обратно в результате реакции)
(V-изменяется обратно в результате реакции) тогда
тогда 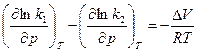
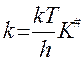 , логарифм. и дифф. по реакции:
, логарифм. и дифф. по реакции: или
или 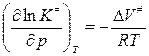 (Vизменяется обратно активности)
(Vизменяется обратно активности) (если ΔV<0- константа скорости увеличивается с ростом давления)
(если ΔV<0- константа скорости увеличивается с ростом давления) k(0)- константа скорости при нулевом давлении.
k(0)- константа скорости при нулевом давлении.