|
|
Клинико-генеологический методТеоретическая часть. Клинико-генеологический метод был предложен в 1865 году Ф. Гальтоном. Метод основан на прослеживании интересующего нас признака (нормального или патологического) в семье, с указанием родственных связей между отдельными членами этой семьи (составлением родословной). Клинико-генеалогический метод дает возможность: - выявлять наследственный характер признака; - определять тип наследования; - определять зиготность членов родословной; - определять особенности взаимодействия генов; - устанавливать сцепленное наследование и проводить картирование хромосом; - определять пенетрантность гена; - изучать закономерности мутирования отдельных генов; - устанавливать носительство мутантного гена тем или иным членом семьи; - определять вероятность генетически обусловленных событий и рассчитывать риск наследования патологического гена (признака) при медико-генетическом консультировании. Клинико-генеалогический метод часто осложняется невозможностью сбора достаточного количества информации из-за малодетности семей, либо из-за прерывания связей между поколениями, отсутствия связей между родственниками, либо по морально-этическим причинам. Клинико-генеалогический метод лежит в основе медико-генетического консультирования и включает 3 этапа: 1 этап – клиническое обследование; 2 этап – составление родословной; 3 этап – генетический анализ родословной.
Первый этап – клиническое обследование. При составлении родословной сбор сведений о семье начинается с человека, которого называют пробанд (обычно это больной с изучаемым заболеванием или признаком). В сведениях о пробанде указывается анамнез заболевания, включающий начальные признаки и возраст их манифестации, последующее течение болезни; если пробанд – ребенок – сведения о раннем психомоторном и последующим умственном и физическом развитии. Чем больше поколений удается проследить и чем более полно охватить членов родословной при сборе сведений, тем больше вероятность получения достоверных сведений о характере наследования изучаемого признака. Сбор генетической информации проводится путем опроса, анкетирования, личного собеседования. Опрос начинается обычно с родственников по материнской линии. В родословную вносят сведения о выкидышах, абортах, мертворожденных, бесплодных браках, внебрачных детях и др. При сборе генетической информации о проявлении изучаемого признака ведется краткая запись данных о каждом члене рода с указанием его родства по отношению к пробанду. Обычно указывается фамилия (для женщин девичья фамилия), имя, отчество, дата рождения и смерти. Полученные данные записываются в медико-генетическую карту. При сборе информации необходимо внимательно анализировать сообщения об инфекциях и травмах, следует учитывать гетерогенность и варьирующую экспрессивность наследственных заболеваний. Необходимо выяснять акушерский анамнез, учитывать наличие и характер профессиональных вредностей, возраст, национальность, место жительства семьи, профессию, наличие хронических заболеваний в семье, причину смерти умерших и др. На основании изученных данных составляется анамнез (греч. – anamnesis – воспоминание).
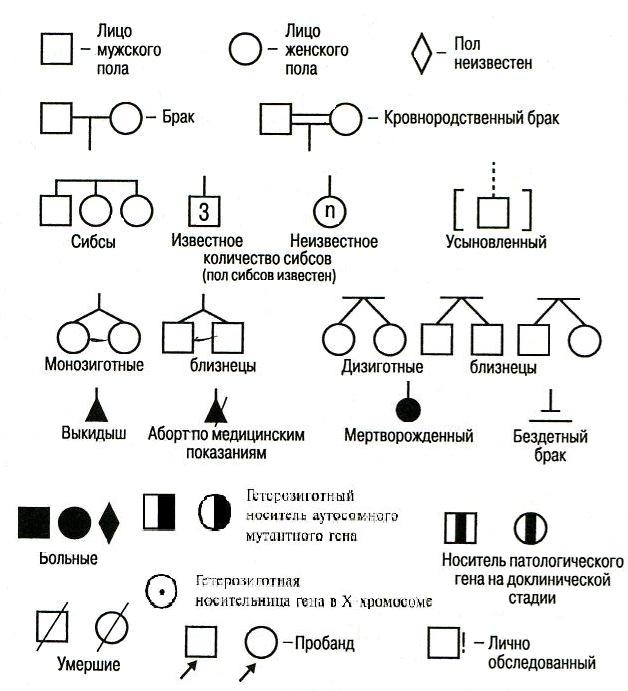
Второй этап – составление родословной. После сбора сведений составляется графическое изображение родословной, для этого используется система символов, предложенная в 1931 году Г. Юстом. (Рис. 1). При составлении графического изображения родословной важно соблюдать следующие правила: 1. Составление родословной начинают с пробанда. Братья и сестры (сибсы) располагаются в порядке рождения слева направо, начиная со старшего. 2. Все члены родословной располагаются строго по поколениям, в один ряд. 3. Поколения обозначаются римскими цифрами слева от родословной сверху вниз. 4. Арабскими цифрами нумеруется потомство одного поколения (одного ряда) слева направо. Благодаря такой нумерации каждый член семьи имеет свой шифр (например: I-1, I-2, II-2, II-4 и др.) 5. Указывается возраст членов семьи (родословной), в связи с тем, что некоторые болезни проявляются в разные периоды жизни. 6. Отмечаются лично обследованные члены родословной. Графическое изображение родословной может быть вертикально-горизонтальным или расположенным по кругу (в случае многочисленных данных). Схема родословной сопровождается описанием обозначений под рисунком (легендой).
Рис. 1 Символы, используемые при составлении родословных
Третий этап – генетический анализ родословной. Этот этап требует хороших знаний критериев установленных типов наследования. Задача генетического анализа – установление наследственного характера заболевания и типа наследования, выявление гетерозиготных носителей мутационного гена, установление генотипа пробанда и, как заключение, прогнозирование потомства. Анализ родословной рекомендуется проводить в следующей последовательности: 1. Установление, является ли данный признак (заболевание) наследственным. Если признак встречается несколько раз в разных поколениях (имеет семейный характер), то можно предполагать, что признак имеет наследственную природу. 2. Определение типа наследования признака. Для этого учитывают: - во всех ли поколениях и как часто среди членов родословной встречается признак; - одинакова ли частота признака у обоих полов и если нет, то у какого пола встречается чаще; - детям какого пола передается признак от больного отца и от больной матери; - есть ли семьи, в которых от больных родителей рождаются здоровые дети, или наоборот, от здоровых родителей рождаются больные дети; - какая часть потомства имеет наследуемый признак в семьях, где болен один из родителей. Наряду с заболеваниями, обусловленными ядерными генами у человека все более изученными становятся заболевания митохондриальные и болезни геномного импритинга. Под генетическим импритингом понимают эпигенетический процесс, приводящий к стойким функциональным различиям экспрессии гомологичных генов, полученных от одного из родителей. Так, например, в настоящее время хорошо известно влияние ряда отцовских и материнских генов на вес плода, степень развития плаценты и другие особенности внутриутробного развития. Наличие генов, подверженных импритингу, четко установлено для хромосом 7,11,15. Предполагают, что такие гены присутствуют и в хромосомах 2, 3, 6, 14 и 20. В настоящее время идентифицировано не менее 30 импритированных генов, часто группирующихся в кластеры, по некоторым данным, таких генов в геноме человека насчитывается не менее 100. Большой класс составляют болезни экспансии тринуклеотидных повторов. и наследственные болезни обмена. У человека установлены следующие типы наследования, подчиняющиеся менделеевским закономерностям наследования: аутосомно-рецессивное наследование (АР), аутосомно-доминантное наследование (АД). Болезни с нетрадиционными типами наследования: доминантное, сцепленное с Х хромосомой наследование (ХД), рецессивное, сцепленное с Х хромосомой наследование (ХР), сцепленное с Y хромосомой наследование (YН) и цитоплазматическое наследование (ЦН). В зависимости от типа наследования родословные выглядят по-разному.
Аутосомно-доминантный тип наследования (АД). При аутосомно-доминантном типе наследования мутантный ген локализован в аутосоме и проявляется в генотипах АА и Аа. Для этого типа наследования характерны: равная вероятность встречаемости данного признака как у мужчин, так и у женщин. Признак прослеживается при достаточном по численности потомстве в каждом поколении по вертикали. - признак передается одинаково от больных родителей (как матери, так и отца) к детям (Рис. 2); - у здоровых родителей рождается здоровое потомство; - у больных родителей может рождаться здоровое потомство; По аутосомно-доминантному типу наследуются полидактимия (шестипалость), брахидактилия (короткопалость), ахондроплазия (карликовость), синдром Марфана (паучьи пальцы), ангиоматоз сетчатой оболочки, метгемоглобинемия (гемоглобины Бостон, Чикаго), аниридия, глаукома, синдром Ван дер Хеве, врожденный вывих бедра, гиперхолистеринемия, глухонемота (некоторые формы), нейрофиброматоз, подагра, анемия серповидно-клеточная (АД тип с неполным доминированием), ахондроплазия и др.
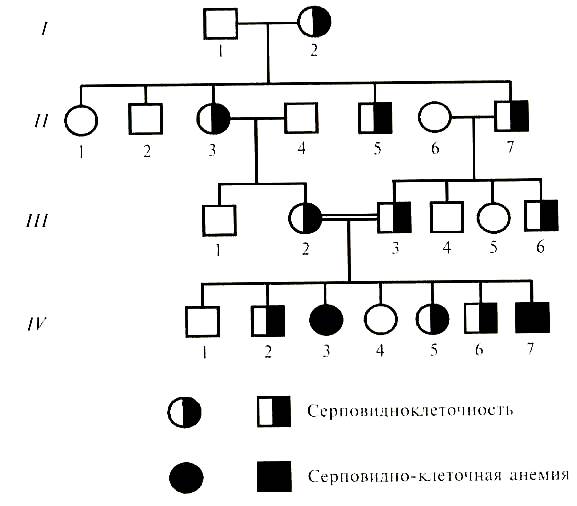
Рис. 2 Родословная семьи с наследованием синдактилии. Аутосомно-рецессивный тип наследования (АР). При АР типе наследования мутантный ген проявляет свое действие только в гомозиготном состоянии (аа). При аутосомно-рецессивном наследовании заболевание (признак0 встречается в родословных редко и не во всех поколениях, при этом вероятность заболевания у мальчиков и девочек одинакова (Рис. 3). Признак может появиться у детей, родители которых здоровы и являлись гетерозиготными носителями мутантного гена. Вероятность появления рецессивного потомства возрастает в близкородственных браках, где оба родителя могут быть носителями одного и того же рецессивного аллеля, полученного от общего предка. Рецессивный признак от родителей, как от отца, так и от матери, передается одинаково. К заболеваниям с аутосомно-рецессивным типом наследования относятся многие болезни обмена веществ, среди которых фенилкетонурия (ФКУ), галактоземия, альбинизм общий, муковисцидоз, акаталазия, алькаптонурия, гидроцефалия, болезнь Вильсона (гепато-церебральная дистрофия), мышечная дистрофия Дюшена (или миопатия), цистинурия болезнь Тея-Сакса и др.
Рис. 3 Родословная семьи с наследованием серповидноклеточности и серповидно-клеточной анемии.
Х-сцепленное рецессивное наследование (ХР). ХР тип наследования характеризуется следующими особенностями: мутантный ген локализован в Х хромосоме и проявляется в генотипе ХаХа у женщин и ХаY у мужчин. - От здоровых родителей, если мать гетерозиготный носитель, могут родиться больные дети – мальчики; больные мужчины не передают заболевание своим сыновьям, но их дочери становятся гетерозиготными носителями болезни; - Больные девочки могут родиться только в семьях, где отец болен, а мать гетерозиготна по мутантному гену (Рис. 4).
Рис. 4 Родословная семьи с наследованием гемофилии. Характерной особенностью родословных при ХР-наследовании является преимущественное проявление признака у гемизиготных (ХаY) мужчин, которые наследуют его от матерей-носителей рецессивного аллеля. Как правило, признак наследуется мужчинами через поколение от деда по материнской линии к внуку. У женщин ХР признак проявляется лишь в гомозиготном состоянии, вероятность чего возрастает в близкородственных браках. По ХР типу наследуются гемофилия, дальтонизм, альбинизм, ангидрозная эктодермальная дисплазия, ангиокератома, ихтиоз врожденный (большинство форм ихтиоза летальны), пигментный ретинит (ХР форма) и др.
Х-сцепленное доминантное наследование (ХД). ХД тип наследования характеризуется следующими признаками: - доминантный мутантный аллель локализован в Х хромосоме и может проявляться как в гомозиготном (ХАХА), в гетерозиготном (ХАХа), так и в гемизиготном (хАу) состоянии; - лица с генотипом ХАХА, ХАХа, ХАY – больны, с генотипом ХаХа, ХаY – здоровы; - болеют как мужчины, так и женщины, однако больных женщин вдвое больше, чем мужчин; - заболевание проявляется в каждом поколении; - если болен отец, то все его дочери будут больны, а сыновья здоровы; - если мать больна, то вероятность рождения больного ребенка 50%, независимо от пола. - больными будут дети только тогда, когда болен один из родителей; - у здоровых родителей все дети будут здоровы (Рис. 5).
Рис. 5 Родословная семьи с наследованием дефекта зубной эмали. При анализе родословных в ряде случаев очень трудно отличить Х-сцепленный доминантный тип от аутосомно-доминантного типа наследования. Наиболее информативны те семьи, в которых передача заболевания происходит через больных отцов. В этом случае наблюдается избирательное поражение потомков женского пола – все дочери таких больных отцов заболевают, т.к. получат от отца Х-хромосому, несущую патологический ген, а все сыновья будут здоровы, т.к. унаследуют от отца Y-хромосому. Х-сцепленный доминантный тип наследования встречается крайне редко. По ХД типу наследуются фолликулярный кератоз, фосфатемия, коричневая окраска эмали зубов, маторно-сенсорная нейропатия 1 Х-типа (ОМIМ: 302800).
Рис. 6 Родословная семьи с наследованием гипертрихоза ушной раковины.
Y-сцепленный (галандрический) тип наследования. Данный тип наследования характеризуется только прямой передачей признака от отца к сыну (Рис. 6). В настоящее время идентифицировано около 100 генов, локализованных в Y-хромосоме. Большинство из них обусловливают развитие организма по мужскому типу, участвуют в сперматогенезе, в контроле роста тела и зубов. Мутации в некоторых генах приводят к развитию рака яичек, простаты и другим гонадопластомам. Все гены Y-хромосомы делятся на 3 группы: 1. Гены псевдоаутосомных областей, идентичные в Х- и Y-хромосомах. Мутации в генах этой группы нарушают конъюгацию геносом (половых хромосом) в мейозе у мужчин, приводят к бесплодию. 2. Группа включает 10 Х-Y-гомологичных генов, локализованных в некомбинирующих областях Yp иYq. Эти гены экспрессируются во многих тканях и органах, включая яички и простату. 3. Группа включает 11 Y-специфичных генов, располагающихся в некомбинирующих областях Yp иYq. Продукты этих генов могут играть роль транскрипционных факторов и цитокининовых рецепторов, выполнять функции протеинкиназ и фосфатаз. Некоторые из этих генов формируют AZF-регион, микроделеции в котором часто приводят к мужскому бесплодию: азооспермии и олигоспермии. Y-сцепленные заболевания, как правило, возникают вследствии мутаций de novo. Мальчики, которые получили от своего отца мутацию, нарушающую развитие и функционирование мужских гонад, оказываются стерильными и не могут передать ее своему потомству, поэтому родословные с этим признаком оказываются неинформативными. В целом, Y-хромосома характеризуется высокой нестабильностью, вследствие наличия большого числа различных повторов, мобильных генетических элементов, аберрантной рекомбинации между гомологичными облястями Х- и Y-хромосом или несбалансированного обмена между сестринскими хроматидами Y-хромасомы. Примерами сцепленного с Y-хромосомой признака является гипертрихоз, синдиктимия. При цитоплазматическом наследовании (ЦН) признак передается по материнской линии через цитоплазму, т.к. гены, обусловливающие цитоплазматическую наследственность расположены в цитоплазматических молекулах ДНК. Так, например, все митохондрии человека имеют материнское происхождение и получены человеком с материнской яйцеклеткой. При наличии мутации в том или ином количестве митохондрий яйцеклетка может передать мутации ДНК митохондрии своим детям обоего пола. Скорость мутаций в митохондриальной ДНК в 17 раз выше, что обусловливает высокую частоту спорадических случаев митохондриальных заболеваний. Цитоплазматическая наследственность не подчиняется законам наследования ядерных генов. Клинические проявления митохондриальных заболеваний характеризуются задержкой физического развития, дисфункцией щитовидной железы, печеночной недостаточностью, миопатиями и кардиомиопатиями и многое другое.
Пенетрантность и экспрессивность. В генетике человека и в медицинской генетике часто выстречаются признаки, которые наследуются по типу неполного доминирования, по типу кодоминирования и по различным типам взаимодействия неаллельных генов. В процессе онтогенеза не все гены реализуются в признак. Некоторые из них оказываются блокированными другими неаллельными генами, проявлению некоторых признаков неспособствуют, неблагоприятствуют внешние условия. Пробиваемость гена в признак называется пенетрантностью. Пенетрантность отражает частоту фенотипического проявления имеющейся в генотипе генетической информации. Пенетрантность выражается в процентах особей, у которых анализируемый аллель фенотипически проявляется. Пенетрантность может быть полной (100%) и неполной (< 100%). При анализе родословной и составлении генетического прогноза необходимо учитывать значение пенетрантности, установленное для данного признака. Например: арахнодактилия наследуется как доминантный аутосомный признак с пенетрантностью 30%. Леворукость – рецессивный аутосомный признак с полной пенетрантностью. Определите вероятность проявления обеих аномалий в семье, где оба родителя гомозиготны по обоим парам генов. Какова вероятность появления детей с арахнодактилией? Решение: А – арахнодактилия, а – нормальная длина пальцев, В – праворукость, в – леворукость. Родители: мать – АаВв – с арахнодактимией, праворукая; отец – АаВв – с арахнодактимией, праворукий.
Анализ потомства
Следовательно, с учетом пенентрантности 30% признака арахнодактилия в семье 5,625% детей будут иметь обе аномалии (18,75%×30%=5,625%). Вероятность появления детей с арахнодактимией составляет 22,55 (16,875%+5,625%=22,5%). Фенотипическое проявление наследственной информации характеризуется показателем, который называется экспрессивность. Экспрессивность характеризует степень выраженности признака и зависит как от дозы соответствующего аллеля гена, так и от факторов среды.
Задание: 1. Изучите конспекты лекций и литературу по данной теме. 2. Зарисуйте в тетради систему символов по Г. Юсту, используемых при составлении родословной. 3. Изучите особенности родословных с различными типами наследования АД, АР, ХД, ХР, YН, ЦН. Зарисовать в тетради родословные с разными типами наследования и провести их анализ. 4. Запишите в тетрадь и выучите примеры заболеваний с разными типами наследования и их клиническое проявление. 5. Постройте родословную для анализа наследования хореи Гентингтона – тяжелого наследственного заболевания (обычно болезнь развивается в возрасте более 40-45 лет), используя следующие данные анмнеза: пробанд – здоровый молодой человек, обратившийся в медико-генетическую консультацию в связи с предполагаемой женитьбой. Отец пробанда болен хореей Гентингтона, а мать здорова и имеет здоровых брата, сестру, отца и мать. У отца пробанда имеется здоровый брат пожилого возраста и сестра, страдающая этим заболеванием; их отец (дедушка пробанда) был здоров, а мать (бабушка пробанда) болела хореей. Известно также, что у бабушки пробанда были две здоровые сестры и два больных брата, а ее отец (прадедушка пробанда) также страдал этим заболеванием. - Определите тип наследования этого заболевания; - Определите возможные генотипы генотипы родителей пробанда, а также других членов родословной; - Рассчитайте вероятность того, что сам пробанд является носителем патологического гена и у него появятся признаки заболевания при достижении соответствующего возраста (пенетрантность патологического гена 100%); - Составьте генетический прогноз потомства пробанда в различных вариантах брака при предполагаемом генотипе пробанда Аа (♀АА х♂ Аа;. ♀Аа х ♂Аа; ♀аа х ♂Аа). Вероятность появления больного потомства выражается в %. 6. Проанализируйте родословную на Рис.4, составленную для семьи, в которой были больные гемофилией. Определите тип наследования патологического признака в семье. Укажите членов семьи, которые являются вероятными носителями мутантного гена. Определите вероятность рождения больных детей в браке индивидуума III-7 и здоровой девушки из семьи, в которой никогда не наблюдалась эта болезнь. Определите вероятность в % рождения больных детей и их пол в случае близкородственного брака индивидуумов III-3 и III-9. 7. Составьте и пронумеруйте родословную схему для анализа миопатии Дюшенна (резко выраженная мышечная слабость, развивающаяся в детском возрасте, которая обычно приводит к ранней смерти), используя следующие данные. Пробанд – здоровая женщина, имеющая двух здоровых дочерей и сына, который страдает миопатией. Муж пробанда здоров, два его брата, сестра и родители тоже здоровы. Старший брат пробанда умер в детстве от миопатии, а другой брат здоров и имеет двух здоровых сыновей и здоровую дочь. Сестра пробанда здорова, но имеет сына, больного миопатией, а также здорового сына и здоровую дочь. Родители пробанда здоровы. Определите тип наследования патологического признака (болезни) в семье. Укажите генотипы больных индивидуумов (на схеме можно сделать запись генотипа рядом с символическим изображением соответствующего индивидуума). Определите членов семьи, являющихся гетерозиготными носителями мутантного гена, и обозначьте их на схеме в соответствии с рекомендациями, приведенными на Рис. 1. Установите вероятность в % того, что следующий ребенок, который может родиться у пробанда, будет больным (здоровым). Какова вероятность в % того, что больной ребенок окажется мальчик (девочка)? Рассчитайте вероятность того, что из двух мальчиков, которые могут родиться у пробанда, оба будут больными (оба будут здоровыми). Какова вероятность того, что первый из двух мальчиков будет больным, а второй здоровым (либо наоборот, первый будет здоровым, а второй больным)?. 8. Проведите анализ и постройте генетический прогноз. У человека полидактилия контролируется доминантным геном. Его аллель обусловливает нормальное развитие конечностей. Какова вероятность полидактилии у будущих детей (пенетрантность доминантного гена в данной семье 90%), если: а) один из супругов гетерозиготен, а у второго нормальнее число пальцев; б) оба супруга гетерозиготны. 9. Определите вероятность ретинобластомы (ретинобластома – аутосомно-доминантный признак с неполной пенетрантностью, пенетрантность доминантного гена в одних семьях составляет 60%, в других – 90%) среди детей в семьях, где: а) один из родителей страдает ретинобластомой (пенетрантность в семье 90%); б) один из родителей страдает ретинобластомой (пенетрантность гена в семье 60%); в) один из родителей был оперирован по поводу ретинобластомы и в настоящее время здоров (пенетрантность гена в семье 60%); г) оба родителя здоровы, но отец одного из них умер от ретинобластомы (пенетрантность гена в семье 60%). 10. Дайте генетический прогноз. Витилиго наследуется как аутосомно-доминантный признак с неполной пенетрантностью. Предположим, что пенетрантность доминантного гена в семье 70%. Какова вероятность рождения ребенка с витилиго, если: а) у одного из родителей имеется витилиго; б) оба родителя здоровы, но их первый ребенок имеет витилиго? 11. Определите вероятность рождения в семье детей с различными формами цистинурии, если: у человека одна из форм цистинурии контролируется мутантным аллелем. У гомозигот по данному аллелю наблюдается образование цистиновых камней в почках, у гетерозигот – повышенное содержание цистина в моче. а) у родителей повышенное содержание цистина в моче; б) один из родителей был оперирован по поводу цистиновых камней в почках и в настоящее время здоров, а у второго повышенное содержание цистина в моче; в) один из родителей был оперирован по поводу цистиновых камней в почках и в настоящее время здоров, второй страдает цистинурией. 12. Какова вероятность рождения в семье где мужчина с I группой крови, страдающий подагрой, женился на здоровой женщине, имеющей II группу крови. а) здорового ребенка со II группой крови; б) больного ребенка со II группой крови; в) здорового ребенка с I группой крови; г) больного ребенка с I группой крови? 13. Синдром Марфана наследуется как аутосомно-доминантный признак с перентрантностью 30%. Резус-отрицательность – аутосомно-рециссивный признак с полной пенетрантностью. Определите вероятность рождения в семье, где оба родителя гетерозиготны: а) резус-положительного здорового ребенка; б) резус-положительного больного ребенка; в) резус-отрицательного здорового ребенка; г) резус-отрицательного больного ребенка. 14. Постройте и проанализируйте родословные по заданию преподавателя. 15.Постройте родословную своей семьи. Оформите работу как самостоятельную на отдельных листах в соответствии с планом: 1. Название: Родословная моей семьи. Изучение наследования _______ (назовите изучаемый признак). 2. Анамнез (составьте анамнез). 3. Условные обозначения по Г. Юсту (приведите все условные обозначения, которые встречаются в литературе при изучении данной темы. 4. Схема родословной (со всеми общепринятой нумерацией поколений и каждого члена родословной). 5. Анализ родословной, установление типа наследования. Дайте обоснование. 6. Генетический прогноз. Для этого установите возможные генотипы родственников пробанда и самого пробанда. Постройте генетический прогноз для потомства пробанда. 7. Примеры и клиническое проявление болезней с таким же типом наследования. 8. Список использованной литературы.
Вопросы для самоконтроля 1. Что изучает генеалогический метод. 2. Каковы возможности и недостатки генеалогического метода. 3. Как построить родословную. 4. Как провести генетический анализ родословной. 5. К каким типам наследования относятся наследственные признаки человека. Приведите примеры. 6. Как характеризуются родословные с разными типами наследования. 7. Объясните понятие пенетрантность и экспрессивность. Приведите примеры.
Форма отчета: 1) Представление на проверку тетради 2) Представление на проверку родословной своей семьи и устная защита работы.
Цитогенетический метод Теоретическая часть. Цитологический метод основан на микроскопическом изучении хромосом в клетках человека. Цитогенетический метод широко применяется с 1956 года, когда Дж. Тио и Л. Леван установили, что в кариотипе человека 46 хромосом. Цитогенетический метод основывается на данных о хромосомах. В 1960 году на научной конференции в Денвере была принята классификация идентифицируемых хромосом, в соответствии с которой им были даны номера, увеличивающиеся по мере уменьшения размеров хромосом. Эта классификация была уточнена на конференции в Лондоне (1963) и Чикаго (1966). Применение цитогенетического метода позволяет изучать нормальную морфологию хромосом и кариотипа в целом, определять генетический пол организма, и, главное, диагностировать различные хромосомные болезни, связанные с изменением числа хромосом или с нарушением структуры хромосом. Цитогенетический метод позволяет изучать процессы мутагенеза на уровне хромосом и кариотипа. Метод широко применяется в медико-генетическом консультировании для целей пренатальной диагностики хромосомных болезней. В соматических клетках человека диплоидный набор хромосом, 2n=46, а в половых – гаплоидный n=23. При оплодотворении диплоидный набор хромосом восстанавливается. В хромосоме выделяют короткое (р) и длинное (q) плечи. Концы обоих плеч хромосомы называют теломерами. В метафазе митоза хромосомы представлены двумя сестринскими хроматидами, соединенными центромерой. В центромере содержится вещество – кинетохор, участвующее в формировании нитей веретена при клеточном делении. При изучении кариотипа определяют следующие морфометрические характеристики хромосом: Lа – абсолютная длина хромосомы в мкм; Lр – длина короткого плеча; Lg – длина длинного плеча. Iв – плечевой индекс, Iс – центромерный индекс, Lr – относительная длина хромосомы, Ih - процент гетерохроматиновой зоны, Is – индекс спирализации. По значению плечевого индекса определяется форма хромосом. При Iв 1-1,9 хромосома называется равноплечей (метацентрической), 2-4,9 – слабонеравноплечей (субметацентрической), 5 и более – акроцентрической или резко неравноплечей. Для кариотипирования подбирают метафазные пластинки в количестве не менее 30 с одинаковым индексом спирализации. На основании различий в длине выделены 23 пары хромосом. По форме в кариотипе человека имеются метацентрические, субметацентрические и акроцентрические хромосомы. Отнесение хромосом к той или иной группе производится на основе расчета центромерного индекса. На основании размеров и комбинации плечевого и центромерного индексов хромосомы человека в соответствии с Международной Денверской классификацией (1960) сгруппированы в 7 групп, обохзначаемых буквами английского алфавита: A, B, C, D, E, F, G.
Таблица 1 Международная денверская классификация хромосом человека (1960г.)
В настоящее время для идентификации хромосом в соответствии с номенклатурой ISCN-1995 (парижская номенклатура) все чаще используется дифференциальное окрашивание, которое на хромосомах дает полосы поперечной исчерченности, благодаря которым можно более точно идентифицировать пары гомологов. Анализ кариотипа проводят в культуре делящихся соматических и половых клеток. Наиболее часто используют культуру клеток переферической крови, прежде всего лимфоцитов, костного мозга и фибробластов. Для анализа кариотипа плода используют различные клеточные культуры; их выбор определяется сроком беременности (до 12 недель – используют клетки ворсин хориона, в более поздние сроки – клетки плода, выделенные из амниотической жидкости, пуповинной крови и плаценты). Цитологический анализ включает три основынх этапа: 1) Культивирование клеток; 2) Окраска препарата; 3) Микроскопический анализ препарата.
Культивирование. Образец помещают в питательную солевую среду с добавлением цельной сыворотки крупного рогатого скота и белка бобовых растений – фитогемагглютинина, стимулирующего процесс деления клеток. Для увеличения числа метафазных клеток (кариотип изучают в метафазных клетках, где хромосомы достигают наибольшей спирализации и наиболее четко проявляется их форма) за 1,5 часа до окончания культивирования в культуру вводят колхицин (C22H25NO6), который разрушает клеточное веретено, приостанавливает деление клеток на стадии метафазы и увеличивает конденсацию (спирализацию) хромосом. Обычно культивирование составляет 72 часа. После этого клетки отделяют центрифугированием и помещением в гипотонический раствор хлорида калия или цитрата натрия. В гипотонической среде происходит разрыв ядерной оболочки и межхромосомных связей и хромосомы свободно перемещаются в цитоплазму. Затем производится фиксация клеток в фиксаторе Карнуа (3:1): 3 части составляет 96% этиловый спирт ректификат, 1 часть ледяная уксусная кислота. После фиксации клеточную суспензию раскладывают на обезжиренные, охлажденные влажные предметные стекла и высушивают на воздухе.
Окраска. Наиболее простой метод окраски хромосом – это сплошная по Гимза. Сплошная окраска применяется для определения количества хромосом, выявления геномных мутаций и анеуплоидии. Для выявления структурной перестройки хромосом (хромосомные мутации) используют дифференциальную окраску, в результате которой хромосомы приобретают поперечную исчерченность. Расположение и длина темных и светлых полос строго индивидуальна для каждой хромосомы, благодаря этому можно провести более точную идентификацию гомологичных пар и выявить перестройки хромосом. Наиболее эффективен G-метод дифференциального окрашивания, для этого можно использовать краситель Гимзы, после предварительной обработки хромосом раствором трипсина. При таком окрашивании количество полос на хромосомах в метафазных пластинках достигает 400. Для дифокраски используют также R-метод, и Q-метод. После окраски объект заключают в Канадский бальзам, препарат становится постоянным и может храниться десятки лет.
Микроскопирование препаратов метафазных хромосом. Для описания кариотипа человека используется универсальная схема и специальные символы. Например, запись 46,хх – обозначает нормальный кариотип женщины, а 46, ху – нормальный кариотип мужчины. В ряде случаев при изучении хромосом обнаруживают полиморфизм, который наиболее характерен для акроцентрических хромосом и, как правило, отражает вариабельность размеров гетерохроматиновых сегментов, наличие спутников, спутничных нитей в области коротких плеч и их величину. В таблице 2 приведены некоторые из них.
Таблица 2 Обозначение полиморфизма хромосом человека
Установлено, что наличие нормального полиморфизма хромосом увеличивает риск рождения ребенка с хромосомными аномалиями. Среди супружеских пар, у которых наблюдалось рождение детей с пороками развития, а также страдающих бесплодием и привычным невынашиванием беременности, чаще выявляется носительство хромосом с крупными гетерохроматиновыми блоками. Пр   ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|