
|
|
В. Лечение отдельных проявлений СКВ1) При антифосфолипидном синдроме для профилактики тромбозов назначают аспирин в низких дозах, хлорохин, гидроксихлорохин и другие препараты, угнетающие агрегацию тромбоцитов. Кортикостероиды снижают титр антифосфолипидных антител, но не уменьшают риск тромбозов. Если с помощью антиагрегантов не удается предотвратить тромбоз, пожизненно назначают варфарин. Изредка (обычно при беременности) больным назначают гепарин. 2) При тромбоцитопении применяют даназол, 200—600 мг/сут внутрь, в тяжелых случаях — нормальный иммуноглобулин для в/в введения, 500 мг/кг/сут в течение нескольких суток. 3) При поражении кожи, когда неэффективны другие средства, применяют ретиноиды, например изотретиноин, 20—80 мг/сут внутрь, в сочетании с солнцезащитными средствами или дапсоном, 50—200 мг/сут внутрь, или клофазимином, 100 мг/сут внутрь. Дапсон назначают после исключения недостаточности Г-6-ФД. VI. Перекрестный синдром и смешанное заболевание соединительной ткани А. Клиническая картина. Для смешанного заболевания соединительной ткани характерно сочетание симптомов склеродермии, ревматоидного артрита, полимиозита и СКВ. Около 10% больных СКВ удовлетворяют критериям смешанного заболевания соединительной ткани, разработанным Американской ревматологической ассоциацией. Обычно смешанное заболевание соединительной ткани больше всего напоминает склеродермию. Артриты и артралгия отмечаются у 96% больных, отек кистей — у 88%, синдром Рейно — у 84%, нарушение моторики пищевода — у 77%, миозит — у 72%, увеличение лимфоузлов — у 68%, лихорадка, серозит, гепатоспленомегалия — у 20—33% больных. Поражение почек характерно для детей, у взрослых встречается редко. Б. Лабораторные исследования. Характерен высокий титр антител к рибонуклеопротеиду (см. гл. 15, п. II.Д.2.а) и отсутствие или низкий титр антител к другим экстрагируемым ядерным антигенам и ДНК. При исследовании антинуклеарных антител методом иммунофлюоресценции наблюдается пятнистое окрашивание срезов тканей (см. гл. 15, п. II.Г.2.в). У некоторых больных имеются признаки нескольких аутоиммунных заболеваний, но, в отличие от больных смешанным заболеванием соединительной ткани, отсутствуют антитела к рибонуклеопротеиду. В этом случае, если имеющиеся признаки удовлетворяют критериям сразу нескольких аутоиммунных заболеваний, ставят диагноз перекрестного синдрома, а если нет — диагноз недифференцированного заболевания соединительной ткани. Впоследствии обычно появляются признаки, позволяющие поставить диагноз того или иного заболевания: ревматоидного артрита, СКВ, системной склеродермии и т. д. В. Лечение зависит от клинической картины и направлено на преобладающие проявления заболевания. VII. Полимиозит А. Эпидемиология. Полимиозит — аутоиммунное заболевание, сопровождающееся поражением скелетных мышц. Заболевание может начаться в любом возрасте, однако чаще всего — в 50—70 лет (ревматоидный артрит и СКВ развиваются обычно в более молодом возрасте). Около 70% больных — женщины. Предполагается, что в патогенезе заболевания участвуют T-лимфоциты и антитела к тРНК-синтетазам (см. гл. 15, п. II.Д.5) — ферментам белкового синтеза, которые обеспечивают присоединение аминокислоты к тРНК. Б. Клиническая картина. Существует несколько классификаций полимиозита. Согласно одной из них, выделяют 5 форм заболевания: 1. Первичный полимиозит. Заболевание начинается внезапно или постепенно. Характерны слабость проксимальных мышц, миалгия, атрофия мышц. У 15% больных наблюдается выраженная артралгия, у 7% заболевание начинается с лихорадки, озноба и синдрома Рейно. В последнем случае обычно ставится неверный диагноз. Первыми чаще всего поражаются мышцы голеней. Больные жалуются на то, что с трудом встают со стула. Затем присоединяется поражение мышц шеи, плеч и бедер и возникает дисфагия — больному трудно глотать жидкость, и она выливается через нос. Дисфагия часто приводит к аспирации пищи. У детей возможна дыхательная недостаточность. Кортикостероиды при этой форме полимиозита умеренно эффективны, в 25—50% случаев требуется назначение иммунодепрессантов (см. гл. 15, п. VII.Г.2). 2. Первичный дерматомиозит. Начало заболевания такое же, как при первичном полимиозите. Однако для дерматомиозита характерна сыпь красного или лилового цвета вокруг глаз, на скулах и над межфаланговыми суставами (папулы Готтрона). В более тяжелых случаях сыпь распространяется на область коленных, локтевых и голеностопных суставов, а также на шею и верхнюю часть спины в виде шали. На поздних стадиях заболевания возникают шелушение, атрофия кожи, возможен некроз. Поражение кожи может предшествовать или сопутствовать поражению мышц, но практически никогда не встречается без полимиозита. 3. Полимиозит при злокачественных новообразованиях. Злокачественные новообразования у мужчин старше 40 лет часто сопровождаются полимиозитом, поэтому при развитии заболевания в этом возрасте требуется тщательное обследование. Чаще всего полимиозит встречается при раке легкого, предстательной железы, желудка, толстой кишки, яичников и молочной железы. Иногда полимиозит возникает при лимфомах. Кортикостероиды неэффективны, улучшение наступает после излечения основного заболевания. 4. Детский полимиозит почти всегда сопровождается сыпью и миалгией (более выраженной, чем у взрослых). Заболевание обычно неуклонно прогрессирует. Прогноз менее благоприятный, чем при других формах полимиозита. Часто развиваются атрофия, обызвествление и оссификация мышц, контрактуры. Характерная особенность детского дерматомиозита — васкулиты и тромбозы с поражением ЖКТ, проявляющиеся болью в животе, изъязвлением слизистой, желудочно-кишечным кровотечением и даже перфорацией кишечника. 5. Полимиозит при других аутоиммунных заболеваниях: синдроме Шегрена, смешанном заболевании соединительной ткани, СКВ и ревматоидном артрите. 6. Миозит с включениями — редкая форма полимиозита — проявляется слабостью дистальных мышц. При биопсии пораженных мышц обнаруживаются включения в мышечных волокнах. Активность мышечных ферментов повышена незначительно. Кортикостероиды при этой форме полимиозита неэффективны. В. Диагностика. Диагноз ставится на основании характерной клинической картины, повышения активности КФК и альдолазы в сыворотке, результатов ЭМГ и биопсии мышц. Однако полимиозит нельзя исключить даже в отсутствие характерных лабораторных признаков заболевания. Трудности диагностики полимиозита приводят к тому, что сразу поставить диагноз удается лишь у 20% больных. 1. ЭМГ выявляет изменения, характерные для поражения как нервов — спонтанные фибрилляции, повышенная возбудимость при введении электродов и положительные потенциалы, — так и мышц — низкоамплитудные полифазные потенциалы действия двигательных единиц 2. Гистологическое исследование. Выявляются дегенерация, вакуолизация и некроз мышечных волокон. Поражаются преимущественно волокна, расположенные по периферии мышечных пучков. Регенерирующие мышечные волокна имеют базофильную цитоплазму и центрально расположенные ядра. В мышечной ткани и стенках сосудов нередко обнаруживаются инфильтраты, состоящие из лимфоцитов и нейтрофилов. Для дерматомиозита характерны более выраженные периваскулярные инфильтраты с большим количеством B-лимфоцитов. Иногда при дерматомиозите наблюдается картина, характерная для васкулитов. 3. Серологические исследования в диагностике полимиозита малоинформативны. У небольшой части больных выявляется ревматоидный фактор, у 20% — антинуклеарные антитела. Диагностическое значение имеют антитела к антигенам Pm-1, Scl-70 и Jo-1, однако их находят лишь у немногих больных (см. гл. 15, п. II.Д.5). Г. Лечение 1. Назначают преднизон, 60 мг/сут внутрь, или другой кортикостероид в эквивалентной дозе. По мере улучшения состояния дозу постепенно снижают. За несколько недель до повышения мышечной силы снижаются активность КФК и альдолазы, а также СОЭ. Поддерживающие дозы преднизона в большинстве случаев составляют 10—20 мг/сут внутрь. Для уменьшения риска побочных действий кортикостероиды назначают через день. 2. Если после 6 нед лечения кортикостероидами состояние не улучшается или заболевание продолжает прогрессировать спустя 3 нед после начала лечения, назначают метотрексат, 15 мг/нед внутрь, постепенно увеличивая ее до 50 мг/нед. Метотрексат в дозе более 20 мг/нед обычно вводят парентерально. К препаратам второго ряда относится азатиоприн, 2—3 мг/кг/сут внутрь. При лечении азатиоприном ежемесячно проводят общий анализ крови и каждые 3 мес определяют биохимические показатели функции печени. При неэффективности лечения и после исключения злокачественных новообразований назначают циклофосфамид, циклоспорин, хлорамбуцил или комбинацию этих препаратов. VIII. Склеродермия и склеродермические состояния — группа заболеваний, для которых характерно повышенное содержание коллагена в дерме и сужение просвета мелких сосудов. Развитию этих заболеваний способствуют наследственная предрасположенность, контакт с некоторыми химическими веществами и инфекции. Патогенез склеродермии окончательно не изучен, однако для его объяснения предложено несколько механизмов: 1) повторное повреждение эндотелия приводит к поражению мелких сосудов и развитию синдрома Рейно; 2) активная пролиферация фибробластов приводит к синтезу избыточных количеств коллагена и уплотнению кожи; 3) повышение активности T-хелперов и функциональная недостаточность T-супрессоров способствуют синтезу аутоантител. А. Классификация. В настоящее время существует несколько классификаций склеродермии и склеродермических состояний. Согласно одной из них, выделяют следующие клинические формы заболевания. Ограниченная склеродермия а. Линейная склеродермия обычно начинается в детском возрасте. Для нее характерно появление участков утолщенной кожи в виде длинных полос на руках и ногах или на коже лба и волосистой части головы («сабельный удар»). При гистологическом исследовании пораженных участков кожи обнаруживают фиброз и лимфоцитарную инфильтрацию дермы. При лабораторном исследовании могут выявляться признаки, характерные для системной склеродермии. Поражение может приводить к контрактурам, которые иногда требуют хирургического лечения. Прогноз в основном благоприятный, в большинстве случаев заболевание заканчивается самостоятельно, лишь у незначительной части больных (менее 5%) развивается системная склеродермия. б. Бляшечная склеродермия. При этой форме склеродермии на коже появляются желтоватые или восковидные пятна или бляшки, окруженные розовым или лиловым ободком, которые со временем уплотняются. Лимфоцитарная инфильтрация в пораженных участках кожи при бляшечной склеродермии выражена больше, чем при других формах заболевания. При обострениях назначают пеницилламин (см. гл. 15, п. III.Г.3.д). Бляшечная склеродермия редко прогрессирует и обычно заканчивается самостоятельно. 2. Системная склеродермия. Критерии системной склеродермии, принятые Американской ревматологической ассоциацией, приведены в табл. 15.10. а. CREST-синдром. Термин представляет собой аббревиатуру, составленную из первых букв следующих слов: C alcinosis — обызвествление, R aynaud's phenomenon — синдром Рейно, E sophageal dysmotility — нарушение моторики пищевода, S clerodactyly — склеродактилия, T eleangiectasia — телеангиэктазия. Для CREST-синдрома характерно достаточно благоприятное течение. В редких случаях наблюдается тяжелый синдром Рейно и выраженная дисфагия. б. Собственно системная склеродермия — самая тяжелая форма склеродермии, протекает с поражением внутренних органов, в течение 10 лет умирает более половины больных. 3. Диффузный эозинофильный фасциит обычно развивается подостро. Заболеванию часто предшествует физическая нагрузка. Характерны болезненность, припухлость и уплотнение кожи кистей, предплечий, стоп и голеней. Уплотнение и рубцовые изменения подкожной клетчатки приводят к контрактурам и синдрому запястного канала. Иногда поражаются туловище и лицо. Для диффузного эозинофильного фасциита, в отличие от системной склеродермии, синдром Рейно нехарактерен. При гистологическом исследовании в коже, фасциях и мышцах выявляются клеточная инфильтрация и фиброз, в воспалительном инфильтрате много эозинофилов. При исследовании крови обнаруживают эозинофилию и гипергаммаглобулинемию. Обычно через несколько месяцев или лет заболевание заканчивается самостоятельно. Применение кортикостероидов в умеренных дозах (преднизон, 10—20 мг/сут внутрь, или другие кортикостероиды в эквивалентных дозах) позволяет уменьшить выраженность симптомов. Некоторые авторы к диффузному эозинофильному фасцииту относят синдром эозинофилии—миалгии (см. гл. 8, п. III.А.5), который возникает после приема триптофана, содержащего токсичные примеси, а также заболевание, подобное системной склеродермии, которое возникает при употреблении плохо очищенного рапсового масла (его вспышка была зарегистрирована в начале 80-х гг. в Испании). 4. Склеродермические состояния, вызванные лекарственными средствами и химическими веществами. Поражение кожи и подкожной клетчатки, аналогичное склеродермии, наблюдается при: 1) применении блеомицина, 2) трансплантации костного мозга; 3) протезировании молочной железы силиконовыми протезами, 4) контакте с профессиональными вредностями (поливинилхлоридом, трихлорэтиленом и другими веществами). Стандартные схемы лечения склеродермии при склеродермических состояниях не всегда эффективны.  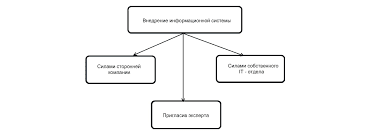 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор...  Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|