
|
|
Нарушения синтеза гема. ПорфирииПорфирии - гетерогенная группа заболеваний, вызванная нарушениями синтеза гема вследствие дефицита одного или нескольких ферментов. Классификации порфирий Единой классификации порфирий нет. Порфирии делят по причинам на: 1) Наследственные. Возникают при дефекте гена фермента, участвующего в синтезе гема; 2) Приобретенные. Возникают при ингибирующем влиянии токсических соединений (гексохлорбензол, соли тяжелых металлов - свинец) на ферменты синтеза гема. В зависимости от преимущественной локализации дефицита фермента (в печени или эритроцитах) порфирин делится на: 1) печеночные – наиболее распространенны. К ним относятся острая перемежающаяся порфирия (ОПП), поздняя кожная порфирия, наследственная копропорфирия, мозаичная порфирия; 2) эритропоэтические – врожденная эритропоэтическая порфирия (болезнь Гюнтера), эритропоэтическая протопорфирия. В зависимости от клинической картины, порфирии делят на: 1) острые. 2) хронические. Негативные последствия порфирий связаны с дефицитом гема и накоплением в организме промежуточных продуктов синтеза гема – порфириногенов и продуктов их окисления. При эритропоэтических порфириях порфирины накапливаются в нормобластах и эритроцитах, при печёночных — в гепатоцитах. Для каждого вида порфирии существует определенный уровень ферментативного дефекта, в результате накапливаются продукты, синтезирующиеся выше этого уровня. Эти продукты являются основными диагностическими маркерами заболевания. Порфириногены ядовиты, при тяжёлых формах порфирий они вызывают нейропсихические расстройства, нарушения функций РЭС и повреждения кожи. Нейропсихические расстройства при порфириях связаны с тем, что аминолевулинат и порфириногены являются нейротоксинами. В коже на солнце порфириногены легко превращаются в порфирины. Кислород при взаимодействии с порфиринами переходит в синглетное состояние. Синглетный кислород стимулирует ПОЛ клеточных мембран и разрушение клеток, поэтому порфирии часто сопровождаются фотосенсибилизацией и изъязвлением открытых участков кожи. Порфириногены бесцветны и не флуоресцируют, а порфирины проявляют интенсивную красную флуоресценцию в ультрафиолетовых лучах. Избыток порфиринов который выводиться с мочой, придает ей темный цвет («порфирин» в переводе с греч. означает пурпурный). При лёгких формах наследственных порфирий заболевание может протекать бессимптомно, но приём лекарств, являющихся индукторами синтеза аминолевулинатсинтазы, может вызвать обострение болезни. В некоторых случаях симптомы болезни не проявляются до периода полового созревания, когда повышение образования β-стероидов вызывает индукцию синтеза аминолевулинатсинтазы. Порфирии наблюдают и при отравлениях солями свинца, так как свинец ингибирует аминолевулинатдегидратазу и феррохелатазу. Некоторые галогенсодержащие гербициды и инсектициды являются индукторами синтеза аминолевулинатсинтазы, поэтому попадание их в организм сопровождается симптомами порфирии. Виды порфирий Острая перемежающая порфирия (ОПП) – причина – дефект гена, кодирующего ПБГ – дезаминазу. Наследуется по аутосомно-доминатному типу. Происходит накопление ранних предшественников синтеза гема: 5- АЛК (5- ALA) и порфобилиногена (ПБГ). Бесцветный ПБГ на свету превращается в порфибилин и порфирин, они предают моче темный цвет. АЛК оказывает нейротоксическое действие, приводя к вялому параличу конечностей и парезу дыхательной мускулатуры. Последнее вызывает острую дыхательную недостаточность. Заболевание проявляется в среднем возрасте, провоцируется приемом анальгетиков, сульфаниломидных препаратов, так как они увеличивают синтез АЛК – синтазы. Клинической симптоматикой являются острые боли в животе, рвота, запор, сердечно-сосудистые нарушения, нервно-психические расстройства. Не наблюдается повышенной чувствительности к свету, так как метаболическое нарушение проходит на стадии, предшествующей образованию уропорфириногена. Для лечения применяют препарат нормосанг – аргинат гема. Действие основано на том, что гем, по механизму отрицательной обратной связи блокирует трансляцию АЛК – синтазы, а, следовательно, падает синтез АЛК и ПБГ, чем и достигается купирование симптоматики. Врожденная эритропоэтическаяпорфирия - редкое врожденное заболевание, наследуемое по аутосомно-рецессивному типу. Для нее характерна низкая активность уропорфириноген-III-косинтазы и высокая - уропорфириноген-I-синтазы. Образование уропорфириногена-I значительно превосходит синтез уропорфириногена-III (нормального изомера на пути синтеза гема). Хотя генетическое нарушение распространяется на все клетки, проявляется оно по неизвестной причине преимущественно в эритропоэтической ткани. Пациенты экскретируют с мочой большие количествауропорфириногена-I и копропорфириногена-I; которые на воздухе самопроизвольно окисляются в уропорфирин-I и копропорфирин-I — красные флуоресцирующие пигменты. Циркулирующие эритроциты содержат большое количество уропорфирина-I, однако, наивысшая концентрация этого порфирина отмечена в клетках костного мозга. Отмечается светочувствительность кожи, обусловленная активацией ПОЛ при действии света на порфириновые соединения. У пациентов отмечаются трещины на коже, часто наблюдаются гемолитические явления. Наследственная копропорфирия — аутосомно-доминантное нарушение, обусловленное дефицитом копропорфириногеноксидазы — митохондриального фермента, ответственного за превращение копропорфириногена III в протопорфириноген IX. Копропорфириноген III в больших количествах удаляется из организма в составе фекалий и с мочой. Копропорфириноген на свету и воздухе быстро окисляется, превращаясь в красный пигмент копропорфирин. Ограниченная способность к синтезу гема (особенно в стрессовых условиях) приводит к дерепрессии АЛК-синтазы. В результате наблюдается избыточное образование АЛК, порфобилиногена и других интермедиатов синтеза гема. У пациентов обнаруживаются все признаки и симптомы, связанные с избытком АЛК и порфобилиногена, которые характерны для перемежающейся острой порфирии, имеется повышенная светочувствительность, обусловленная присутствием избыточных количеств копропорфириногенов и уропорфириногенов. Мозаичная порфирия, или наследственная фоторопорфирия, является аутосомно-доминантным нарушением, при котором происходит частичное блокирование ферментативного превращения протопорфириногена в гем. В норме это превращение осуществляется двумя ферментами, протопорфириногеноксидазой и феррохелатазой, локализованными в митохондриях. Содержание протопорфириногеноксидазы составляет лишь половину нормального количества. У пациентов с мозаичной порфирией наблюдается относительная недостаточность содержания гема в стрессовых условиях, а также дерепрессированное состояние печеночной АЛК-синтазы. Что ведет к перепроизводству всех интермедиатов синтеза гема на участках перед заблокированной стадией. Пациенты с мозаичной порфирией экскретируют с мочой избыточные количества АЛК, порфобилиногена, уропорфирина и копропорфирина, а с фекалиями выделяют уропорфирин, копропорфирин и протопорфирин. Моча больных пигментирована и флуоресцирует, а кожа чувствительна к свету так же, как и у больных поздней кожной порфирией (см. ниже). Поздняя кожная порфирия является наиболее распространенной формой порфирии. Обычно она связана с поражениями печени, особенно при избыточном потреблении алкоголя или перегрузке ионами железа. Вероятной причиной является частичная недостаточность уропорфириноген-декарбоксилазы. Нарушение, по-видимому, передается как аутосомно-доминантный признак, но генетическая пенетрантность различна и в большинстве случаев зависит от наличия нарушений функций печени. Моча содержит повышенные количества уропорфиринов типа I и III; в то же время экскреция с мочой АЛК и порфобилиногена наблюдается сравнительно редко. Иногда моча содержит весьма значительное количество порфиринов, придающих ей розоватый оттенок; при подкислении она чаще всего дает в ультрафиолетовой области розовую флуоресценцию. Печень содержит большие количества порфиринов и поэтому сильно флуоресцирует, тогда как у эритроцитов и клеток костного мозга флуоресценция отсутствует. Главным клиническим проявлением при поздней кожной порфирии является повышенная светочувствительность кожи. У больных не наблюдается ни повышенной активности АЛК-синтазы, ни соответственно избыточного содержания в моче порфобилиногена и АЛК; это коррелирует с отсутствием острых приступов, характерных для перемежающейся острой порфирии. Протопорфирия, или эритропоэтическая протопорфирия, по-видимому, обусловлена доминантно наследуемой недостаточной активностью феррохелатазы в митохондриях всех тканей; клинически эта болезнь проявляется как острая крапивница, вызываемая воздействием солнечных лучей. Эритроциты, плазма и фекалии содержат повышенные количества протопорфирина IX, а ретикулоциты (незрелые эритроциты) и кожа (при исследовании с помощью биопсии) часто флуоресцируют красным светом. Печень, вероятно, тоже вносит вклад в повышение образования протопорфирина IX, однако экскреции с мочой порфиринов и их предшественников не наблюдается.
Синтез гемоглобина Синтезированный в митохондриях гем индуцируется синтез цепей глобина на полирибосомах. Гены цепей глобина расположены в 11 и 16 хромосоме. Цепи глобина формируют глобулы и соединяются с гемом. 4 глобулы нековалентно соединяются в гемоглобин. Гемоглобин начинает синтезироваться на стадии базофильного эритробласта, а заканчивается у ретикулоцитов. В ретикулоцитах также идет синтез пуринов, пиримидинов, фосфатидов, липида. Чувствительным биохимическим индикатором для отличия ретикулоцитов от зрелых клеток является утрата последними глутаминазы. Глутамин в ретикулоцитах - источник углерода для синтеза порфирина и азота для синтеза пурина. Строение гемоглобина Гемоглобин - тетрамерный хромопротеин, имеет массу 64,5кДа, состоит из 4 гемов и 4 глобинов. Глобины представлены полипептидными цепями различных типов a, b, g, d и т.д. a-цепь содержит 141 АК, а b - цепь – 146 АК. Отдельные участки полипептидных цепей образуют правозакрученные a-спирали, особое расположение в пространстве которых формирует глобулы. Глобулаb-субъединицы содержит 8 a-спиралей, а a-субъединицы –7. Гем располагается в щелях между Е и F спиралями глобина, прикрепляясь через гистидин F8 к спирали F с помощью 5 координационной связи железа. Гидрофобные остатки аминокислот окружающие гем, препятствуют окислению железа водой. 4 глобулы с участием гидрофобных, ионных и водородных связей формируют шарообразный тетрамер гемоглобина. Максимально прочные связи, в основном за счет гидрофобных связей, образуются между a- и b-глобулами. В результате образуются 2 димера a1b1 и a2b2. Димеры соединяются между собой в основном полярными (ионными и водородными) связями, поэтому взаимодействие димеров зависит от рН. Димеры легко перемещаются друг относительно друга. В центре тетрамера глобулы прилегают друг к другу неплотно, образуя полость. Функции гемоглобина · Обеспечивают перенос кислорода от легких к тканям. В сутки около 600литров; · Участвует в переносе углекислого газа и протонов от тканей к легким; · Образует гемоглобиновый буфер, регулирует КОС крови.
 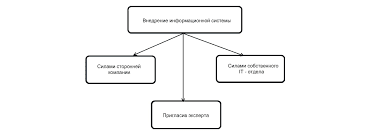 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между... 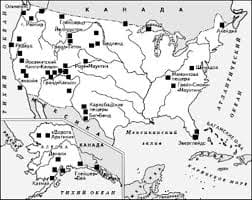 Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|