
|
|
Методы изучения патофизиологииПатофизиологи используют 4 основные группы методов: 1. Эксперимент на живых объектах осуществляется на различных видах животных, на отдельных органах, тканях, клетках и субклеточных структурах. Патофизиологи издавна стремятся воспроизводить на животных типовые патологические процессы (воспаление, лихорадка, аллергия и т.д.), отдельные симптомы и синдромы заболеваний (артериальную гипер– и гипотензию, клонические судороги, почечную и печеночную недостаточность), а также адекватные модели отдельных заболеваний человека (пневмонии, атеросклероза, инфаркта миокарда, некоторых инфекционных заболеваний и т.д. Для постановки эксперимента и познания закономерностей возникновения и течения патологических процессов необходимы: а) рабочая гипотеза или идея; б) метод, обеспечивающий возможность проверки гипотезы; в) правильная оценка результатов эксперимента. Рабочая гипотеза является отправной точкой любого эксперимента. Гипотеза – это попытка объяснить какой–либо процесс на основании имеющихся в данное время фактических данных, результатов. Исключительно важное значение при выполнении эксперимента имеет методика его постановки, т.е. правильный выбор животного, форма осуществления эксперимента (острый или хронический опыт), использование соответствующих приборов, инструментария и правильная организация контрольных опытов. экспериментальные язвы желудка следует вызывать у крыс, а не у кроликов – у них часто наблюдаются спонтанные язвы желудка. Инфекционные процессы не следует изучать на крысах – они устойчивы к инфекции (лучше всего на кроликах, мышах). Аллергию, анафилактический шок лучше всего моделировать на морских свинках, неврозы – на собаках, опухолевый процесс – на мышах. . Для изучения патологических процессов используют следующие основные методы эксперимента: – метод выключения – удаление или повреждение какого–либо органа (хирургическое, химическое, механическое) и сравнение появившихся симптомов с клинической картиной заболевания при предполагаемом поражении функции того же органа у человека (удаление поджелудочной железы, введение аллоксана или дитизона – сахарный диабет); – метод включения – введение в организм животных различных веществ, экстрактов из тканей, гормонов и сравнение этих результатов с результатами аналогичных воздействий при тех или иных заболеваниях человека (введение тиреоидных гормонов – симптомы тиреотоксикоза); – метод раздражения – путем различных воздействий изменяют функции того или иного органа (например, при раздражении блуждающего нерва возникает брадикардия); – метод изолированных или "переживающих" органов (изолированное сердце, печень, легкие и т.д.) позволяет оценить истинный характер, глубину повреждения этого органа и его вклад в развитие недостаточности кровообращения, пищеварения, дыхания и т.п.; – метод парабиоза – соединение и совместное функционирование двух животных (парабионтов), что позволяет выяснить ряд вопросов о гуморальной или нервной природе различных воздействий на организм; – метод тканевых культур (эксплантации) позволяет изучать процессы малигнизации и оценивать эффективность противоопухолевых препаратов; – метод сравнительной патологии – изучение в сравнительном (эволюционном) аспекте лихорадки, воспаления, гипоксии и т.д. Используются и другие методы исследования на животных. 2. Типы передачи наследственных болезней, характеристика, примеры. Типы передачи генных наследственных болезней. Различают аутосомно-доминантный, аутосомно-рецессивный, кодоминантный, промежуточный, смешанный и сцепленный с полом типы передачи наследственных заболеваний. При аутосомно-доминантном типе наследования патологический ген, находящийся в одной из аутосом, доминирует над нормальным ге- ном-аллелем и проявляетсебя в гетерозиготном состоянии (А'а), при этом пенетрантность гена приближается к 100% (неполная пенетрантность доминантного гена встречается редко). При генеалогическом исследовании больные выявляются в каждом поколении. По аутосомно-доминантному типу наследуется болезнь Альцгей- мера — прогрессирующее слабоумие, начинающееся после 40 лет и длящееся от 1 года до 15 лет. Характеризуется выраженной дегенерацией коры, гиппокампа и ствола головного мозга. Патогенез заболевания связан с наличием патологического гена в 21 -й хромосоме.Происходят дегенерация и гибель нейронов, нарушается синтез ацетилхолинтрансферазы в нейронах и резко уменьшается синтез ацетилхолина, нарушается синаптическая передача, постепенно присоединяется нарушение потребления нервными клетками кислорода и глюкозы, ухудшается кровоснабжение мозга. Болезнь характеризуется быстрым или медленным, но неуклонным прогрессированием расстройств памяти, распадом интеллекта, эмоциональной нестабильностью, утратой условных рефлексов и другими неврологическими расстройствами. Хорея Гентингтона развивается у мужчин — носителей патологического гена в 4-й хромосоме. Характеризуется гибелью ГАМК-эргических нейронов и преобладанием дофаминергических нейронов. Проявляется подергиванием мышц, нарушением психики. У женщин из-за усиливающего влияния гена одной изХ-хромосом на ген, находящийся в 4-й хромосоме (феномен геномного импринтинга), заболевание не проявляется. Ахондроплазия — аутосомно-доминантное заболевание, характеризующееся нарушением развития конечностей в длину, повышенной ломкостью костей (размеры туловища нормальные, интеллект сохранен). По аутосомно-доминантному типу наследуются также болезнь Вил- лебранда (нарушение гемостаза), нейрофиброматоз Реклингхаузена, поликистоз почек, полилоз толстой кишки, миотония Томсена, семейная гиперхолестеринемия, брахи-, син- и полидактилия и другие заболевания. При аутосомно-рецессивном типе наследования патологический ген проявляет себя только в гомозиготном состоянии (а'а — носитель гена, здоровый; а'а' — больной). При генеалогическом исследовании больные выявляются не в каждом поколении. Примеры фе- нилкетонурия, альбинизм, алкаптонурия, галактоземия, галассемия, а также гомоцистинурия, гипофизарная карликовость, микроцефалия, некоторые формы шизофрении и эпилепсии, гепатолентикулярная дегенерация (болезнь Коновалова—Вильсона), наследственная юношеская эмфизема легких (дефицит фермента а-1-антитрипсина — антагониста протеолитических ферментов) и многие другие заболевания. При кодоминантном типе наследования проявляют себя оба гена (патологический рецессивный и нормальный). Например, у гетерозиготных носшелей гена серповидно-клеточной анемии в крови обнаруживаются эритроциты с нормальным гемоглобином и гемоглобином типа S. Фено- типически заболевание может проявиться только при дефиците кислорода, когда аномальный гемоглобин кристаллизуется и происходит гемолиз эритроцитов. При нормальном содержании кислорода гемолиз эритроцитов не наблюдается. При промежуточном типе наследования ни один ген не доминирует. При смешанном типе ген, ответственный за развитие патологического признака, проявляет себя у одного больного как доминантный, а у другого — как рецессивный. Наследование, сцепленное с полом, характеризуется тем, что патологический ген находится в половой хромосоме — чаще в Х-хромосоме и может быть как рецессивным, так и доминантным. По этому типу наследуются такие заболевания, как дальтонизм, мышечная дистрофия Дюшен- на, сидеробластная анемия (из-за нарушения обмена порфиринажелезо не встраивается в гем и снижается синтез гемоглобина), фавизм (из-за дефицита глюкозо-6-фосфатдегидрогеназы после приема сульфаниламидов происходит гемолиз эритроцитов), почечный несахарный диабет (нечувствительность рецепторов канальцев почек к АДГ), а также гемофилия А (нарушение гемостаза, обусловленное отсутствием VIII фактора свертывания крови). У женщины — носителя рецессивного гена гемофилии (Х'Х) заболевание не проявляется, так как во 2-й Х-хромосоме есть нормальный аллель, подавляющий ген гемофилии. У мужчины из-за отсутствия аналогичного аллеля в Y-хромосоме наличие гена гемофилии в Х-хромосоме (X'Y) приводит к развитию гемофилии. Хромосомные болезни- Хромосомная болезнь может возникнуть в результате мутаций в гаметах родителей ил1/ в клетках эмбриона на стадии дробления зиготы. Синдром Шерешевского—Тернера (Х-моносомия) характеризуется наличием 44 аутосом + Х0 и отсутствием полового хроматина (телец Барра) в ядрах клеток. У женщин с этим синдромом выявляются низкий рост, широкая короткая шея (часто с характерными крыловидными складками), врожденные пороки сердца, множественные пигментные пятна, недоразвитие молочныхжелез и яичников, первичная аменорея и бесплодие, умственное развитие нормальное. Синдром Y-моносимии (44 + Y0) — организм нежизнеспособен. Синдром Трипло-Х (44 аутосомы + XXX) фенотипически может не проявляться или, наоборот, характеризуется задержкой физического, полового и умственного развития (слабоумие, шизофрения). Часто обнаруживаются высокое твердое небо и эпикант (поперечная складка кожи у внутреннего угла глаза). В ядрах клеток таких женщин имеется два тельца Барра. Синдром Клайнфельтера (44 аутосомы + XXY, или + XXXY и т.п.). Для мужчин с таким синдромом характерны высокий рост, астеническое телосложение евнухоидного типа, гинекомастия, атрофия яичек и бесплодие, часто остеопороз, возможно гомосексуальное и асоциальное поведение. В отличие от нормальных мужчин в ядрах клеток у них обнаруживается половой хроматин (телец Барра столько, сколько лишних Х-хромосом). Синдром Дауна в 94 % случаев характеризуется трисомией в 21-й паре аутосом (45 аутосом + XX у девочек или + XY у мальчиков). В остальных случаях может быть транслокационный вариант хромосомной аномалии (например, перенос фрагмента 21 -й хромосомы на 13-ю или на 14-ю, или на 15-ю, или на 22-ю пары). Для этого синдрома характерны олигофрения разной степени выраженности (имбицильность, дебильность или идиотия), низкий рост, разболтанность суставов, мышечная гипотония, короткие пальцы (может быть синдактилия), поперечная «обезьянья» складка на ладони, монголоидный разрез глаз, эпикантус, увеличенный язык, уменьшение размеров мозга, часто встречаются пороки сердца и аномалии других внутренних органов, недоразвитие половых признаков. Для этих больных характерны также резкое снижение клеточного и гуморального иммунитета, повышенный риск развития лейкоза и формирование ранней катаракты. Синдром Патау (трисомия по 13-й паре аутосом) характеризуется микроцефалией, аномикрофтальмом, деформацией ушных раковин, расщелиной верхней губы и нёба, полидактилией, недоразвитием обеих челюстей, пороками сердца. Синдром Эдвардса (трисомия по 18-й паре аутосом) проявляется деформацией ушных раковин, узкими глазными щелями, гипоплазией нижней челюсти, микроцефалией, пороками сердца, почек и органов пищеварения. Следует заметить, что для всех аутосомных анеуплоидий характер- натриада: множественные пороки развития органов, олигофрения и значительное сокращение продолжительности жизни.
  Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем... 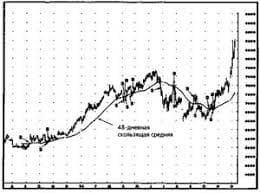 Что вызывает тренды на фондовых и товарных рынках Объяснение теории грузового поезда Первые 17 лет моих рыночных исследований сводились к попыткам вычислить, когда этот... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|