
|
|
Д. Тромбоцитопеническая пурпура, вызванная переливанием эритроцитарной массы1. Клиническая картина. Тромбоцитопеническая пурпура — редкое осложнение переливания эритроцитарной массы. Она проявляется внезапной тромбоцитопенией, кровотечениями из слизистых и петехиями, которые возникают через 7—10 сут после переливания. Диагноз основан на данных анамнеза. Эта форма тромбоцитопенической пурпуры чаще всего встречается у многорожавших женщин и лиц, перенесших многократное переливание эритроцитарной массы. По механизму развития она сходна с тромбоцитопенией новорожденных, вызванной материнскими антителами. Тромбоцитопеническая пурпура, вызванная переливанием эритроцитарной массы, возникает у лиц, у которых отсутствует антиген Zwa. Показано, что этот антиген входит в состав гликопротеида IIb/IIIa. Переливание эритроцитарной массы с примесью тромбоцитов, несущих антиген Zwa, приводит к появлению антител к этому антигену. Полагают, что они перекрестно реагируют с гликопротеидом IIb/IIIa собственных тромбоцитов больного. Лечение а. Переливание тромбоцитарной массы не проводят, поскольку оно обычно неэффективно. Кроме того, донорами тромбоцитарной массы при этом заболевании могут быть лишь 2% людей, у которых тромбоциты не несут антиген Zwa. б. Преднизон, 1—2 мг/кг/сут внутрь, уменьшает геморрагический синдром и повышает число тромбоцитов. в. Заболевание проходит самостоятельно после того, как кровь больного освобождается от тромбоцитов донора. г. Впоследствии для переливания должна использоваться эритроцитарная масса доноров, у которых отсутствует антиген Zwa. IV. Иммунная нейтропения. Число исследований, посвященных иммунным механизмам повреждения нейтрофилов, относительно невелико. Это объясняется отсутствием простых и надежных методов выявления антител к нейтрофилам. Антитела, вызывающие их агглютинацию, обнаруживаются у многорожавших женщин и лиц, перенесших многократное переливание эритроцитарной массы. Выявление этих антител способствовало открытию антигенов HLA и положило начало типированию тканей. Антитела к нейтрофилам обычно определяют в реакции агглютинации. Этот метод более трудоемок, чем реакция гемагглютинации, требует более тщательной очистки клеток, применения специальных сред, строгого соблюдения условий культивирования клеток (например, поддержание постоянной температуры). Другой способ выявления антител к нейтрофилам основан на оценке фагоцитоза нейтрофилов, предварительно инкубированных с исследуемой сывороткой. А. Нейтропения новорожденных, вызванная материнскими антителами, обусловлена иммунизацией матери нейтрофилами плода. Заболевание может возникать у первенцев. Обычно оно проявляется при инфекции. Легкие формы протекают бессимптомно. Диагностика а. Общий анализ крови. Число лейкоцитов может быть нормальным, повышенным или слегка сниженным, однако число нейтрофилов заметно ниже нормы. Иногда они вообще отсутствуют. Отмечается умеренный моноцитоз, иногда — эозинофилия. б. Число клеток в костном мозге повышено, однако сегментоядерные, а иногда и палочкоядерные нейтрофилы отсутствуют. в. Отсутствие антител к антигенам NA1, NA2 и NB1 нейтрофилов (см. гл. 16, п. IV.Б) в сыворотке больных исключает диагноз аутоиммунной нейтропении. 2. Лечение. Нейтропения обычно сохраняется в течение 2—17 нед (в среднем 7 нед). Обычно проводят поддерживающее лечение. Если присоединяется инфекция, назначают антимикробные средства. Кортикостероиды неэффективны. Б. Аутоиммунная нейтропения обычно наблюдается при аутоиммунных заболеваниях и часто сочетается с аутоиммунными гемолитической анемией и тромбоцитопенией. Исключают другие причины нейтропении. Выявление антител к антигенам нейтрофилов NA1, NA2 и NB1 подтверждает диагноз. Назначают кортикостероиды, как при аутоиммунной гемолитической анемии. V. Иммунные коагулопатии. Система гемостаза включает в себя два звена: клеточное (тромбоциты) и гуморальное (факторы свертывания крови). В данном разделе рассматриваются диагностика и лечение иммунных коагулопатий (иммунные тромбоцитопении описаны в гл. 16, п. III). Для коагулопатий характерны гемартрозы, массивные межмышечные и забрюшинные гематомы, желудочно-кишечные кровотечения. Коагулопатии делятся на врожденные и приобретенные. Врожденные коагулопатии обусловлены отсутствием или дефектом какого-либо фактора свертывания, приобретенные — снижением синтеза факторов свертывания, вызванным каким-либо заболеванием, или появлением ингибиторов факторов свертывания. Эндогенные ингибиторы факторов свертывания чаще всего направлены против фактора VIII и представляют собой IgG, реже — IgM и IgA. Причина образования эндогенных ингибиторов факторов свертывания неясна. А. Эндогенные ингибиторы фактора VIII Заболевания и состояния, сопровождающиеся появлением в крови эндогенных ингибиторов фактора VIII а. Заместительная терапия фактором VIII при гемофилии A сопровождается появлением ингибиторов этого фактора примерно у 10% больных. Связь между появлением ингибиторов фактора VIII и количеством введенного препарата отсутствует. Следует отметить, что ингибиторы фактора VIII чаще всего наблюдаются у больных тяжелой гемофилией A. В этом случае они образуются вследствие иммунизации экзогенным фактором VIII, в остальных случаях они имеют аутоиммунную природу. б. Эндогенные ингибиторы фактора VIII наблюдаются при аутоиммунных заболеваниях: СКВ, ревматоидном артрите, неспецифическом язвенном колите, болезни Крона. в. Эндогенные ингибиторы фактора VIII могут появляться у здоровых людей, в том числе у женщин в послеродовом периоде. В последнем случае ингибиторы фактора VIII присутствуют в сыворотке обычно в течение нескольких месяцев. 2. Диагностика. Для выявления ингибиторов фактора VIII плазму больного смешивают с нормальной плазмой в разных соотношениях и затем определяют АЧТВ и активность фактора VIII. Изменение показателей отмечается сразу или через 1—2 ч. В качестве контроля определяют активность фактора VIII нормальной плазмы в течение всего времени исследования. За единицу активности ингибитора принимают такое его количество в 1 мл плазмы, которое снижает активность фактора VIII на 50%. Лечение а. В первую очередь лечат основное заболевание. б. При геморрагическом синдроме вводят фактор VIII, 10 000—40 000 ед/сут в/в. Он инактивирует эндогенные ингибиторы. У некоторых больных через 2—4 сут после введения фактора VIII активность ингибиторов повышается, максимальной она становится примерно через 2 нед. в. Если активность ингибиторов фактора VIII высокая (> 5 ед), проводят плазмаферез или обменное переливание крови с последующим назначением фактора VIII в высоких дозах. г. Другая схема лечения позволяет избежать введения фактора VIII. Она сводится к назначению концентрата фактора IX, 70—90 ед/кг в/в, при необходимости введение повторяют через 12—24 ч. Среди осложнений этого метода лечения следует отметить ДВС-синдром, риск которого повышается при заболеваниях печени. Если концентрат фактора IX содержит следы фактора VIII, возможно повышение активности эндогенных ингибиторов последнего. д. Эффективно длительное лечение кортикостероидами и иммунодепрессантами, например циклофосфамидом, в дозе 1,0—1,5 г/м2 в/в каждые 3—4 нед. Циклофосфамид в более низких дозах неэффективен. При гемофилии A кортикостероиды и иммунодепрессанты неэффективны. е. Ингибиторы фактора VIII видоспецифичны, поэтому при угрожающем жизни кровотечении можно применять свиной фактор VIII. Следует учитывать, что повторное назначение этого препарата может осложняться анафилактическими реакциями. ж. У некоторых больных эффективны нормальный иммуноглобулин для в/в введения в высоких дозах, иногда в сочетании с циклофосфамидом. Эта схема лечения не только вызывает быстрое снижение активности ингибиторов фактора VIII, но и длительно подавляет их продукцию. Возможно, этот эффект обусловлен присутствием в нормальном иммуноглобулине для в/в введения антиидиотипических антител, блокирующих ингибиторы фактора VIII. 4. Прогноз. Половина больных, не страдающих гемофилией, умирает от кровотечений, несмотря на интенсивное лечение. Появление ингибиторов фактора VIII снижает продолжительность жизни больных гемофилией. У здоровых ингибиторы фактора VIII нередко исчезают самостоятельно Б. Эндогенные ингибиторы фактора V обнаруживаются очень редко. По частоте выявления они занимают второе место после эндогенных ингибиторов фактора VIII. 1. Эндогенные ингибиторы фактора V появляются в крови в следующих случаях. а. Ингибиторы фактора V обнаруживаются в раннем послеоперационном периоде у больных, получавших стрептомицин. Однако связь между появлением ингибиторов фактора V и применением стрептомицина пока не доказана. б. Их также обнаруживают у здоровых лиц пожилого возраста. Причины появления ингибиторов фактора V в этом случае также неясны. 2. Диагностика. Диагноз ставится, если 1) протромбиновое время остается увеличенным после добавления к плазме больного нормальной плазмы и 2) определяется низкий уровень фактора V. Следует отметить, что кровотечения возникают далеко не у всех больных с эндогенными ингибиторами фактора V. Лечение а. При кровотечении для восстановления дефицита фактора V и нейтрализации его ингибиторов переливают свежезамороженную плазму. Ограниченный объем плазмы, который возможно перелить, часто не позволяет полностью нейтрализовать ингибиторы, присутствующие в крови больного. б. Переливание тромбоцитарной массы. Фактор V в большом количестве содержится в тромбоцитах, поэтому переливание тромбоцитарной массы иногда позволяет нейтрализовать ингибиторы фактора V. в. Обычно ингибиторы фактора V со временем самостоятельно исчезают из крови. В. Ингибиторы других факторов свертывания выявляются исключительно редко. Описаны ингибиторы фактора IX, а также факторов XIII (при лечении изониазидом), X (при AL-амилоидозе) и протромбина (после операций на сердце и при проказе). VI. Парапротеинемии. Парапротеины — это иммуноглобулины, легкие или тяжелые цепи иммуноглобулинов, продуцируемые одним клоном B-лимфоцитов. Диагноз парапротеинемии ставят, если при электрофорезе, иммуноэлектрофорезе или иммунофиксационном электрофорезе сыворотки или мочи (см. гл. 20, п. I.А) выявляются моноклональные антитела. Заболевания, при которых наблюдается парапротеинемия, перечислены в табл. 16.6. В 30% случаев парапротеинемия обусловлена гемобластозами. Парапротеинемии различаются по типу тяжелых (гамма, мю, альфа, дельта, эпсилон) и легких (каппа и лямбда) цепей. Частота отдельных форм парапротеинемий соответствует относительному содержанию этих цепей в норме. Так, парапротеинемия, проявляющаяся высоким содержанием IgG, встречается в 60% случаев, IgM — в 20%, IgA — в 10%, IgD — менее чем в 1%, IgE — крайне редко, легких цепей — в 7% при соотношении каппа/лямбда — 2:1. А. Доброкачественная моноклональная гаммапатия — распространенная парапротеинемия, которая обнаруживается у 1—1,5% лиц старше 50 лет и 3% лиц старше 70 лет. Эта парапротеинемия обычно не прогрессирует и протекает бессимптомно. 1. Диагностика. В сыворотке выявляются моноклональные иммуноглобулины, их уровень обычно менее 3 г%. Уровень остальных иммуноглобулинов не отличается от нормы, число плазматических клеток в костном мозге не превышает 5%, содержание кальция в сыворотке нормальное, кости не поражены. 2. Лечение обычно не требуется, однако из-за повышенного риска гемобластозов больных с этой парапротеинемией необходимо регулярно обследовать. У 47% больных парапротеинемия сохраняется пожизненно (но не является причиной смерти), у 16% развивается миеломная болезнь, у 10% уровень парапротеина увеличивается до значений, превышающих 3 г% в отсутствие опухолевой трансформации, у 3% развивается первичный амилоидоз, у 3% — макроглобулинемия Вальденстрема, у 2% — другие гемобластозы. Гемобластозы развиваются у 17% больных через 10 лет и у 33% — через 20 лет после постановки диагноза доброкачественной моноклональной гаммапатии. Б. Миеломная болезнь — это самый частый из гемобластозов, при которых происходит трансформация плазматических клеток. 1. Клиническая картина зависит от тяжести заболевания. При поражении костей часто отмечаются боль в спине и груди, патологические переломы. Почти у всех больных выявляются анемия и снижение анионного интервала. При обширном поражении костей наблюдается гиперкальциемия, при продукции легких цепей — нарушение функции почек. Прогрессирование болезни приводит к лейкопении и тромбоцитопении. Повышение вязкости плазмы (из-за высокой концентрации моноклональных иммуноглобулинов) приводит к нарушениям зрения, кровоизлияниям в сетчатку, ишемическому поражению ЦНС, инфаркту миокарда и геморрагическому синдрому. Диагностика а. Уровень моноклональных иммуноглобулинов в сыворотке превышает 3 г%. б. С мочой за сутки выделяется более 12 г белка (это легкие цепи моноклональных иммуноглобулинов). в. Число плазматических клеток в костном мозге превышает 30%. г. Уровень нормальных (поликлональных) иммуноглобулинов в сыворотке снижен. д. При рентгенографии выявляется поражение костей. Чаще встречается остеопороз, однако диагностически значимым является очаговое поражение костей. 3. Лечение направлено на увеличение продолжительности жизни, профилактику осложнений и уменьшение выраженности симптомов. Несмотря на лечение, медиана выживаемости составляет 3 года. Прогноз зависит от уровня моноклональных иммуноглобулинов и бета2-микроглобулина, активности ЛДГ, степени гиперкальциемии, анемии и тромбоцитопении, наличия ХПН, а также от типа моноклональных иммуноглобулинов: IgG, IgA, легкие цепи, IgD, моноклональные иммуноглобулины, содержащие легкую цепь каппа, моноклональные иммуноглобулины, содержащие легкую цепь лямбда (перечислены в порядке ухудшения прогноза). а. Химиотерапия. Существует множество схем лечения миеломной болезни. Обычно они включают кортикостероиды, мелфалан, циклофосфамид, винкристин, доксорубицин, производные нитрозомочевины, хлорамбуцил, блеомицин и этопозид. б. Лучевую терапию при миеломной болезни применяют с паллиативной целью — для уменьшения боли при поражении костей, в том числе при патологических переломах. в. Осложнения миеломной болезни — ХПН, гиперкальциемия, инфекции, нейропатия, эндокринные нарушения, повышенная вязкость плазмы — требуют специального лечения. г. Трансплантация костного мозга. Хотя миеломную болезнь считают неизлечимой, полихимиотерапия в сочетании с трансплантацией костного мозга приводит к длительной ремиссии у 20—25% больных (данные Международного общества по трансплантации костного мозга). В. Макроглобулинемия Вальденстрема — заболевание, в основе которого лежит пролиферация клона плазматических клеток, секретирующих моноклональные IgM — макроглобулины. Обычно оно возникает в возрасте 50—70 лет, несколько чаще у мужчин, чем у женщин. У многих больных отмечаются слабость, утомляемость, склонность к кровотечениям (которая может быть обусловлена как нарушением функции тромбоцитов, так и снижением уровня факторов свертывания), увеличение лимфоузлов, селезенки и печени, может повышаться вязкость плазмы. Кости обычно не поражаются. У некоторых больных в крови появляются криоглобулины, в таких случаях основными проявлениями заболевания становятся синдром Рейно и холодовая крапивница. Диагностика а. У большинства больных отмечается анемия. б. Прямая проба Кумбса чаще всего отрицательна. в. При электрофорезе выявляется фракция макроглобулинов (обычно более 3 г%). г. Уровень нормальных иммуноглобулинов иногда снижен. У некоторых больных макроглобулины, подобно холодовым агглютининам и ревматоидному фактору, образуют комплексы с IgG, преципитирующие при низкой температуре. д. В костном мозге преобладают плазматические клетки (более 30%). е. В отличие от миеломной болезни, при макроглобулинемии Вальденстрема обычно наблюдается увеличение лимфоузлов и гепатоспленомегалия, а поражение костей и гиперкальциемия отмечаются редко. 2. Лечение. Заболевание протекает по-разному. Обычно оно медленно прогрессирует. При повышении вязкости плазмы проводят плазмаферез. Применяется химиотерапия с использованием хлорамбуцила и преднизона или циклофосфамида, винкристина и преднизона. Г. Болезнь тяжелых цепей — группа редких заболеваний, для которых характерно появление в сыворотке аномальных тяжелых цепей иммуноглобулинов (участок CH1 отсутствует, Fc-фрагмент нормальный), имеющих моноклональную природу. Известны 4 типа аномальных тяжелых цепей иммуноглобулинов — гамма, альфа, мю и дельта. Аномальные тяжелые цепи связываются с антителами к нативным иммуноглобулинам и не связываются с антителами к легким цепям иммуноглобулинов. По клинической картине заболевания этой группы сходны с лимфомами. При физикальном исследовании выявляются увеличение лимфоузлов и гепатоспленомегалия, при исследовании костного мозга — диффузная инфильтрация лимфоцитами, плазматическими клетками, эозинофилами и макрофагами. На B-лимфоцитах отсутствуют поверхностные иммуноглобулины. Причины заболеваний этой группы неизвестны. 1. Болезнь тяжелых гамма-цепей обычно встречается у людей старше 40 лет, хотя было описано несколько случаев заболевания в возрасте от 12 до 40 лет. Характерны лихорадка, увеличение лимфоузлов и анемия. Уровень нормальных иммуноглобулинов в сыворотке обычно снижен, в то время как уровень аномальных тяжелых цепей иммуноглобулинов превышает 2 г%. Заболевание может протекать по-разному, выживаемость колеблется от нескольких месяцев до нескольких лет. В терминальной фазе заболевания наблюдается значительное повышение числа плазматических клеток в костном мозге, как при плазмоклеточном лейкозе. Эффективных методов лечения этого заболевания нет. 2. Болезнь тяжелых альфа-цепей — наиболее распространенная среди болезней тяжелых цепей. Обычно встречается у людей моложе 50 лет (чаще между 10 и 30 годами). Характерно выраженное увеличение лимфоузлов брыжейки. Инфильтрация слизистой кишечника лимфоцитами и плазматическими клетками и атрофия ворсинок слизистой кишечника приводят к хроническому поносу и синдрому нарушенного всасывания. У некоторых больных эффективна химиотерапия по схемам, применяемым при лимфомах, у других — антимикробная терапия. 3. Болезнь тяжелых мю-цепей встречается редко и почти всегда сопутствует хроническому лимфолейкозу. Увеличение лимфоузлов нехарактерно. В костном мозге выявляются вакуолизированные плазматические клетки. У большинства больных в моче выявляется высокий уровень легких цепей. Диагноз ставится при выявлении аномальных мю-цепей с помощью иммунохимических методов. Лечат основное заболевание — хронический лимфолейкоз. 4. Болезнь тяжелых дельта-цепей встречается исключительно редко. Описан случай заболевания с развитием ХПН, поражением костей и характерной для миеломной болезни картиной поражения костного мозга. При электрофорезе сыворотки между фракциями гамма- и бета-глобулинов обнаруживается белок, который связывается с антителами к тяжелым дельта-цепям, но не связывается с антителами к легким цепям иммуноглобулинов. Д. Криоглобулинемия. Криоглобулины — это иммуноглобулины, преципитирующие при низкой температуре. Для обнаружения криоглобулинов кровь забирают предварительно нагретым шприцем и инкубируют при 37°C до образования сгустка. Отделившуюся сыворотку охлаждают до 4°C и через 3 сут определяют преципитаты. В норме сыворотка содержит не более 80 мкг/мл криоглобулинов, при криоглобулинемии их концентрация достигает 500—5000 мкг/мл. 1. Клиническая картина. К характерным проявлениям относятся синдром Рейно, акроцианоз, сухая гангрена пальцев, геморрагическая сыпь. Она обычно локализуется на ногах, особенно в области голеностопных суставов, где иногда появляются язвы. При переохлаждении наблюдаются артралгия и ограничение подвижности в суставах. Если криоглобулины обладают свойствами холодовых агглютининов, возможен внутрисосудистый гемолиз. К поздним осложнениям относится ХПН. При биопсии почек выявляются отложения иммуноглобулинов и комплемента под базальной мембраной клубочков. 2. Классификация. Типы криоглобулинемии и заболевания, при которых они выявляются, представлены в табл. 16.7. Смешанная и поликлональная криоглобулинемии примерно в половине случаев обусловлены гемобластозами и системными васкулитами. Лечение а. Больному рекомендуют избегать переохлаждения. б. Лечат сопутствующие заболевания. в. В тяжелых случаях показан плазмаферез, хотя он позволяет добиться только временного улучшения. Во избежание преципитации криоглобулинов плазмаферез проводят в теплом помещении — как при болезни холодовых агглютининов (см. гл. 16, п. II.Д.2.в). Единого мнения об эффективности плазмафереза при ХПН и нефротическом синдроме, вызванных криоглобулинемией, нет. г. Для подавления синтеза криоглобулинов иногда применяют циклофосфамид, хлорамбуцил и кортикостероиды. Е. AL-амилоидоз, ранее называвшийся первичным амилоидозом, — заболевание, при котором в тканях откладываются парапротеины, представляющие собой моноклональные легкие цепи иммуноглобулинов. Отложение парапротеинов в тканях сопровождается необратимым повреждением внутренних органов, которое приводит к снижению продолжительности жизни. AL-амилоидоз возникает в отсутствие других заболеваний. 1. Клиническая картина зависит от того, в каких тканях преимущественно откладываются легкие цепи иммуноглобулинов. Часто наблюдаются слабость, утомляемость, головокружение, обмороки, геморрагическая сыпь вокруг глаз, охриплость, дисфагия, отеки ног, одышка, периферическая нейропатия. В 34% случаев обнаруживается гепатомегалия, в 22% — макроглоссия, в 4% — спленомегалия и увеличение лимфоузлов. У половины больных заболевание начинается в 65 лет. Чаще всего поражаются сердце (у 35% больных развивается сердечная недостаточность), язык (20%), ЖКТ (характерны дисфагия и синдром нарушенного всасывания), почки (у 35% развивается нефротический синдром), нервная система (у 17% больных отмечается периферическая и вегетативная нейропатия) и костный мозг (30%). 2. Диагностика. Диагноз AL-амилоидоза основан на клинической картине и результатах лабораторных исследований. а. Электрофорез белков сыворотки и мочи. При электрофорезе (у 40% больных) и иммуноэлектрофорезе (у 68% больных) белков сыворотки определяется фракция моноклональных белков. При электрофорезе белков сыворотки и мочи эта фракция обнаруживается у 89% больных. Примерно у 70% больных эта фракция состоит из моноклональных иммуноглобулинов, у остальных — из моноклональных легких цепей. При AL-амилоидозе, в отличие от миеломной болезни, гиперпродукция каппа-цепей встречается в 2 раза реже, чем гиперпродукция лямбда-цепей. б. Производят биопсию пораженного органа: костного мозга, прямой кишки, почки, удерживателя сгибателей пальцев рук или ног, печени, кожи, икроножного нерва, миокарда, подкожной клетчатки живота. Препараты окрашивают конго красным и исследуют с помощью поляризационного микроскопа. При этом видны двоякопреломляющие нити зеленого цвета. в. Поскольку в 20% случаев AL-амилоидоз сопутствует миеломной болезни, при обследовании больных производят трепанобиопсию крыла подвздошной кости, рентгенографию костей и определение уровня кальция в сыворотке. г. У 82% больных обнаруживается протеинурия, у 5—60% повышен уровень креатинина в сыворотке, у 50% снижен уровень IgG. Анемия встречается лишь при AL-амилоидозе, сопутствующем миеломной болезни. 3. Прогноз. Медиана выживаемости составляет 12 мес и зависит от локализации поражения: при сердечной недостаточности она составляет 6 мес, при вегетативной нейропатии — 9 мес, при нефротическом синдроме — 17 мес, при синдроме запястного канала — 31 мес, при периферической нейропатии — 56 мес. 4. Лечение. Лечения, позволяющего продлить жизнь при амилоидозе, в настоящее время не существует. С паллиативной целью назначают мелфалан и преднизон, иногда в сочетании с колхицином. Это лечение позволяет уменьшить протеинурию и отеки примерно у трети больных с нефротическим синдромом. При поражении других органов лечение, как правило, неэффективно. Трансплантацию почек и сердца не проводят, поскольку смерть наступает от поражения других органов. VII. Иммунное угнетение кроветворения. Угнетение кроветворения, приводящее к панцитопении, может быть обусловлено иммунными механизмами. Самые частые заболевания этой группы — апластическая анемия и парциальная красноклеточная аплазия. А. Апластическая анемия проявляется панцитопенией. Характерно снижение числа ретикулоцитов. При исследовании костного мозга обнаруживается аплазия или тяжелая гипоплазия — число клеток не превышает 10% от нормы. Апластическая анемия возникает вследствие поражения стволовых кроветворных клеток и нарушения их пролиферации и созревания. Апластическую анемию вызывают ионизирующие излучения, цитостатики, вирусы (например, гепатитов A, B и C), некоторые органические (например, бензол) и неорганические соединения. В зависимости от интенсивности действия этих факторов апластическая анемия может быть обратимой или необратимой. Примерно в половине случаев причину заболевания установить не удается. В этом случае говорят о первичной апластической анемии. У больных первичной апластической анемией в крови выявляются T-супрессоры, угнетающие кроветворение in vitro. Вероятно, in vivo они оказывают такое же действие, что косвенно подтверждается следующими наблюдениями: 1) при апластической анемии часто бывает эффективна иммуносупрессивная терапия; 2) иммуносупрессивная терапия способствует приживлению сингенного костного мозга; 3) применение циклофосфамида перед трансплантацией аллогенного костного мозга способствует восстановлению собственного кроветворения. По-видимому, в патогенезе первичной апластической анемии участвуют и гуморальные факторы, поскольку в присутствии сыворотки больных наблюдается угнетение роста культуры нормальных стволовых кроветворных клеток. Роль аутоиммунных механизмов в развитии первичной апластической анемии не ясна. Среди других причин заболевания нельзя исключить врожденные дефекты стволовых клеток, поскольку у некоторых больных после ремиссии, вызванной иммунодепрессантами, впоследствии развиваются миелодиспластические синдромы, у других — пароксизмальная ночная гемоглобинурия или острый миелолейкоз. 1. Диагностика. Начальные симптомы заболевания — утомляемость, одышка при физической нагрузке, лихорадка, кровоточивость. При исследовании крови обнаруживается панцитопения, снижение числа ретикулоцитов, морфология клеток крови не изменена. При исследовании костного мозга клетки кроветворного ростка отсутствуют или их содержание не превышает 10% от нормы, иногда видны скопления эритроидных клеток. Дифференциальная диагностика с миелодиспластическими синдромами и острыми лейкозами основана на результатах исследования костного мозга и цитогенетического исследования. 2. Лечение. Без лечения 90% больных умирают в течение года. а. Трансплантацию аллогенного костного мозга, совместимого по антигенам HLA, проводят после подавления иммунитета высокими дозами циклофосфамида. Для успешной трансплантации у взрослых необходимо, чтобы у донора и реципиента совпадали не менее 5 из 6 антигенов HLA (HLA-A, HLA-B и HLA-DR). Различие более чем по одному из этих антигенов приводит к высокому риску реакции «трансплантат против хозяина» (см. гл. 16, п. VIII и гл. 17). У детей трансплантация может быть удачной при совместимости донора и реципиента по одному гаплотипу. Основные осложнения трансплантации аллогенного костного мозга при апластической анемии — отторжение трансплантата, реакция «трансплантат против хозяина» и длительная иммуносупрессия. Риск этих осложнений ниже у больных, которым перед трансплантацией не переливали эритроцитарную массу. В настоящее время эффективность трансплантации аллогенного костного мозга у детей составляет 80—90%, у взрослых — 40—50%. Единого мнения о тактике лечения больных апластической анемией старше 40 лет — аллогенная трансплантация костного мозга или применение антитимоцитарного иммуноглобулина — нет. Трансплантация костного мозга в этом случае снижает риск миелодиспластических синдромов и острого миелолейкоза, вероятно, вследствие разрушения трансформированных клеток реципиента лимфоцитами донора при развитии реакции «трансплантат против хозяина». б. Антитимоцитарный и антилимфоцитарный иммуноглобулины. Антитимоцитарный иммуноглобулин эффективен примерно у 60% взрослых и 50% детей, больных апластической анемией. Антитимоцитарный иммуноглобулин обычно вводят в дозе 40 мг/кг/сут в/в в течение 5 сут или 20 мг/кг/сут в/в в течение 8 сут. Действие развивается через 3—4 мес. Среди осложнений отмечаются сывороточная болезнь и другие аллергические реакции, оппортунистические инфекции. Сывороточная болезнь возникает через 7—10 сут после введения антитимоцитарного иммуноглобулина и проявляется лихорадкой, пятнисто-папулезной сыпью и артралгией. Поражение почек (вследствие отложения иммунных комплексов) встречается редко. При сывороточной болезни назначают кортикостероиды в высоких дозах. Одновременное назначение антилимфоцитарного иммуноглобулина и циклоспорина эффективно у 70% взрослых больных. Действие развивается быстрее, чем при монотерапии антилимфоцитарным иммуноглобулином. Вначале назначают циклоспорин и антилимфоцитарный иммуноглобулин, 0,75 мл/кг/сут в/в в течение 8 сут, затем только циклоспорин. Длительность применения циклоспорина 6 мес. Дозу циклоспорина (в среднем 6 мг/кг внутрь 2 раза в сутки) подбирают индивидуально, ориентируясь на его концентрацию в сыворотке (примерно 400 нг/мл) и побочные эффекты. Одно из преимуществ этой схемы лечения заключается в том, что циклоспорин предупреждает развитие сывороточной болезни. в. Переливание эритроцитарной и тромбоцитарной массы обычно необходимо на протяжении всего курса лечения. Если запланирована трансплантация костного мозга, то до нее переливаний эритроцитарной и тромбоцитарной массы следует избегать, поскольку они повышают риск отторжения трансплантата. Чтобы инактивировать лимфоциты, предотвратить их приживление и развитие реакции «трансплантат против хозяина», клеточную массу для переливания облучают в дозе 30 Гр. г. Андрогены. При апластической анемии раньше применяли оксиметолон, 0,5—1 мг/кг внутрь 2 раза в сутки (максимальная доза — 100 мг/сут). Однако этот метод лечения малоэффективен. Следует помнить, что при апластической анемии андрогены не могут заменить описанные выше методы лечения. д. Колониестимулирующие и ростовые факторы. Гранулоцитарный и гранулоцитарно-моноцитарный колониестимулирующие факторы — филграстим и молграмостим — и препараты эритропоэтина — эпоэтин альфа и эпоэтин бета — иногда применяют в качестве дополнения к другим методам лечения. Б. Парциальная красноклеточная аплазия. В основе заболевания лежат как гуморальные, так и клеточные иммунные механизмы. Сыворотка больных угнетает эритропоэз в культуре нормальных клеток костного мозга. Хотя сывороточный фактор, угнетающий созревание эритроцитов, не установлен, полагают, что он вырабатывается T-лимфоцитами. 1. Клиническая картина. Сначала появляются одышка при физической нагрузке, утомляемость, головная боль. Вскоре анемия становится столь выраженной, что требует постоянных переливаний эритроцитарной массы. Поскольку парциальная красноклеточная аплазия может быть вторичной, в клинической картине могут преобладать симптомы основного заболевания. Чаще всего это тимома (иногда с миастенией), коллагенозы, неспецифический язвенный колит, болезнь Крона, хронический лимфолейкоз. 2. Диагностика. Парциальную красноклеточную аплазию следует подозревать во всех случаях анемии неясного происхождения, требующей частого переливания эритроцитарной массы. Средний эритроцитарный объем обычно слегка увеличен (100—105 мкм3), морфология эритроцитов не изменена. Отмечается снижение числа ретикулоцитов. Число лейкоцитов и тромбоцитов обычно в норме. При исследовании костного мозга выявляется почти полное отсутствие клеток эритроидного ростка, в остальном картина костного мозга не изменена. Лечение а. Кортикостероиды. У половины больных эффективен преднизон, 1—2 мг/кг/сут внутрь. Эффект проявляется через несколько недель или месяцев после начала лечения. б. Лечение основного заболевания. При тимоме, а иногда и в ее отсутствие эффективна тимэктомия. Лечение коллагенозов, неспецифического язвенного колита и болезни Крона также улучшает состояние больных. в. Спленэктомия неэффективна. г. Циклофосфамид, 50 мг/сут внутрь, или азатиоприн, 1—2 мг/кг/сут внутрь, эффективны у 60% больных. д. Антитимоцитарный и антилимфоцитарный иммуноглобулины (см. гл. 16, п. VII.А.2.б) также вызывают ремиссию. е. Нормальный иммуноглобулин для в/в введения, 400—500 мг/кг/сут в течение 5 сут, может быть эффективным при парциальной красноклеточной аплазии, устойчивой к другим методам лечения. VIII. Реакция «трансплантат против хозяина» — угрожающее жизни состояние, которое развивается после трансплантации аллогенного костного мозга и может приводить к тяжелому поражению внутренних органов. Чаще всего она возникает у больных с иммунодефицитом. Распознавание лимфоцитами донора антигенов реципиента запускает иммунный ответ, в процессе которого клетки реципиента подвергаются атаке цитотоксическими T-лимфоцитами донора. Характерное проявление реакции «трансплантат против хозяина» — тяжелая панцитопения. А. Клиническая картина. Характерна пятнисто-папулезная сыпь на мочках ушей, шее, ладонях, верхней части груди и спины. На слизистой рта образуются язвы, придающие ей вид булыжной мостовой, иногда появляется белый налет, напоминающий кружево. Характерна лихорадка. На ранних стадиях отмечается гипербилирубинемия. Панцитопения сохраняется на всем протяжении заболевания. В тяжелых случаях возникает профузный кровавый понос. Больные умирают от печеночной недостаточности, дегидратации, метаболических нарушений, синдрома нарушенного всасывания, кровопотери и панцитопении. Реакция «трансплантат против хозяина» развивается в следующих случаях. 1. При переливании необлученных компонентов крови при иммунодефиците, например при злокачественных новообразованиях (особенно лимфогранулематозе), первичных иммунодефицитах и больным после трансплантации органов. ВИЧ-инфекция не повышает риск реакции «трансплантат против хозяина». 2. При переливании необлученных компонентов крови, совместимой по антигенам HLA, больным с нормальным иммунитетом реакция «трансплантат против хозяина» возникает редко. Однако описаны случаи реакции «трансплантат против хозяина» посл   Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем... 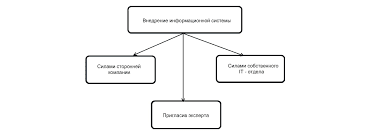 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|