
|
|
Классификация гемолитической анемииНаиболее часто встречающиеся формы гемолитической анемии:
Микросфероцитарная гемолитическая анемия (болезнь Минковского — Шоффара) — наследственное заболевание, обусловленное дефектом белков мембраны эритроцитов, приобретающих сферическую форму с последующим их разрушением макрофагами селезенки. Эпидемиология Этиология и патогенез Классификация Примерная формулировка диагноза: Клиника Анемия нормохромная в большинстве случаев умеренная (гемоглобин 90—100 г/л, при частых и глубоких гемолитических кризах она более выражена (гемоглобин 40—50 г/л), иногда заболевание в течение длительного периода протекает без анемии на фоне небольшого ретикулоцитоза и умеренно выраженной эритробластической реакции костного мозга. Важным признаком является микросфероцитоз эритроцитов. Средний диаметр их бывает меньше 6,3 мкм, средний объем нормальный, средняя толщина заметно увеличена (больше 2,1 мкм). Сфероцитарный индекс (отношение между диаметром и толщиной эритроцита) всегда снижен в среднем до 2,7 (вместо 3,4—3,9 в норме). В период гемолитических кризов значительно увеличивается свободная фракция билирубина сыворотки крови. Продолжительность жизни эритроцитов, меченных Cr, уменьшена почти в 2 раза по сравнению с нормой. Секвестрация их происходит преимущественно в селезенке. Метод кислотных эритрограмм выявляет присущие этой болезни характерные изменения — резкое удлинение времени гемолиза, смещение его максимума вправо. Отмывание эритроцитов от плазмы резко ускоряет гемолиз. При гемолитических кризах наблюдается иногда небольшой лейкоцитоз со сдвигом формулы влево, при явлениях гиперспленизма возможны умеренная лейкопения и тромбоцитопения. Количество ретикулоцитов обычно в пределах 5—10%, гемолитический криз сопровождается увеличением содержания их в несколько раз. В костном мозге резко увеличен эритробластический росток или даже преобладают эритро- и нормобласты. Иногда после гемолитического криза при снижении уровня фолиевой кислоты и витамина В12 обнаруживаются мегалобласты. Выявляющийся параллелизм между сфероцитозом эритроцитов и понижением их осмотической резистентности, а также повышением аутогемолиза, корригируемый глюкозой, не строго специфичен для наследственной микросфероцитарной гемолитической анемии. Верификация диагноза Лечение При наличии тяжело протекающей желчнокаменной болезни наряду со спленэктомией решается вопрос о показаниях к холецистэктомии или к вскрытию и дренированию общего желчного протока. Из гемотерапевтических средств используют трансфузии отмытых или размороженных эритроцитов, при возникновении тромбозов назначаются антикоагулянты. Прогноз благоприятный, однако возможны летальные исходы (инфекции, тромбозы, реже — тяжелые гемолитические кризы).
046. Гемолитические аутоиммунные анемии. Классификация Под названием гемолитические анемии объединяется группа приобретенных и наследственных заболеваний, характеризующихся повышением внутриклеточным или внутрисосудистым разрушением эритроцитов. Аутоиммунные гемолитические анемии включают формы заболевания, связанные с образованием антител к собственным антигенам эритроцитов. Эпидемиология Этиология и патогенез К изоиммунным относятся гемолитические анемии новорожденных, обусловленные несовместимостью по системам АВО и резус между матерью и плодом, посттрансфузионные гемолитические анемии. Классификация Примерная формулировка диагноза: Клиника Анемия носит нормохромный, иногда гиперхромный характер, при гемолитических кризах обычно отмечается выраженный или умеренный ретикулоцитоз. В периферической крови обнаруживается макроцитоз и микросфероцитоз эритроцитов, возможно появление нормобластов. СОЭ в большинстве случаев увеличена. Содержание лейкоцитов при хронической форме бывает нормальным, при острой — встречается лейкоцитоз, достигающий иногда высоких цифр со значительным сдвигом лейкоцитарной формулы влево. Количество тромбоцитов обычно нормальное. При синдроме Фишера — Ивенса аутоиммунная гемолитическая анемия сочетается с аутоиммунной тромбоцитопенией. В костном мозге эритропозз усилен, редко выявляются мегалобласты. У большинства больных снижена осмотическая резистентность эритроцитов, что обусловлено значительным числом микросфероцитов в периферической крови. Содержание билирубина увеличено за счет свободной фракции, повышено и содержание стеркобилина в кале. Неполные тепловые агглютинины обнаруживаются с помощью прямой пробы Кумбса с поливалентной антиглобулиновой сывороткой. При положительном тесте с помощью антисывороток к IgG, IgM и т. д. уточняется, к какому классу иммуноглобулинов относятся выявляемые антитела. Если на поверхности эритроцитов менее 500 фиксированных молекул IgG, проба Кумбса отрицательна. Подобное явление наблюдается обычно у больных с хронической формой аутоиммунной гемолитической анемии или перенесших острый гемолиз. Кумбс-негативными оказываются и случаи, когда на эритроцитах фиксированы антитела, принадлежащие к IgA или IgM (в отношении которых поливалентная антиглобулиновая сыворотка менее активна). Примерно в 50% случаев идиопатических аутоиммунных гемолитических анемий одновременно с появлением иммуноглобулинов, фиксированных на поверхности эритроцитов, выявляются антитела к собственным лимфоцитам. Гемолитическая анемия, обусловленная тепловыми гемолизинами, встречается редко. Для нее характерны гемоглобинурия с выделением мочи черного цвета» чередование периодов острого гемолитического криза и ремиссий. Гемолитический криз сопровождается развитием анемии, ретикулоцитоза (в отдельных случаях тромбоцитоза) и увеличением селезенки. Отмечаются повышение уровня свободной фракции билирубина, гемосидеринурия. При обработке донорских эритроцитов папаином удается обнаружить у больных монофазные гемолизины. У некоторых пациентов оказывается положительной проба Кумбса. Гемолитическая анемия, обусловленная Холодовыми агглютининами (холодовая гемагглютининовая болезнь) имеет хроническое течение. Она развивается при резком повышении титра Холодовых гемагглютининов. Различают идиопатические и симптоматические формы заболевания. Ведущим симптомом болезни является чрезмерно повышенная чувствительность к холоду, которая проявляется в виде посинения и побеления пальцев рук и ног, ушей, кончика носа. Расстройства периферического кровообращения приводят к развитию синдрома Рейно, тромбофлебитов, тромбозов и трофических изменений вплоть до акрогангрены, иногда холодовой крапивницы. Возникновение вазомоторных нарушений связано с образованием при охлаждении крупных внутрисосудистых конгломератов из агглютинированных эритроцитов с последующим спазмом сосудистой стенки. Эти изменения сочетаются с усиленным преимущественно внутриклеточным гемолизом. У части больных встречается увеличение печени и селезенки. Наблюдаются умеренно выраженная нормохромная или гиперхромная анемия, ретикулоцитоз, нормальное количество лейкоцитов и тромбоцитов, увеличение СОЭ, незначительное повышение уровня свободной фракции билирубина, высокий титр полных Холодовых агглютининов (выявляемый методом агглютинации в солевой среде), иногда признаки гемоглобинурии. Характерной является агглютинация эритроцитов in vitro, возникающая при комнатной температуре и исчезающая при подогревании. При невозможности выполнения иммунологических тестов диагностическое значение приобретает провокационная проба с охлаждением (в сыворотке крови, полученной от перетянутого жгутом пальца после опускания его в ледяную воду, определяется повышенное содержание свободного гемоглобина). При холодовой гемагглютининовой болезни в отличие от пароксизмальной холодовой гемоглобинурии гемолитический криз и вазомоторные нарушения возникают только от переохлаждения тела и гемоглобинурия, начавшаяся в условиях холода, прекращается с переходом больного в теплое помещение. Симптомокомплекс, свойственный холодовой гемагтлютининовой болезни, может возникнуть на фоне различных острых инфекций и некоторых форм гемобластозов. При идиопатических формах заболевания полного выздоровления не наблюдается, при симптоматических прогноз зависит главным образом от тяжести основного процесса. Пароксизмальная холодовая гемоглобинурия относится к числу редких форм гемолитических анемий. Ею заболевают люди обоего пола, чаще дети. У больных с пароксизмальной холодовой гемоглобинурией после пребывания на холоде могут появиться общее недомогание, головная боль, ломота в теле и другие неприятные ощущения. Вслед за этим начинается озноб, повышается температура, отмечается тошнота и рвота. Моча приобретает черную окраску. Одновременно иногда выявляются желтушность, увеличение селезенки и вазомоторные нарушения. На фоне гемолитического криза у больных обнаруживают умеренную анемию, ретикулоцитоз, повышение содержания свободной фракции билирубина, гемосидеринурию и протеинурию. Окончательный диагноз пароксизмальной холодовой гемоглобинурии устанавливают на основании обнаруженных двухфазных гемолизинов по методу Доната — Ландштейнера. Для нее не характерна аутоагглютинация эритроцитов, постоянно наблюдающаяся при холодовой гемагтлютинацией ной болезни. Гемолитическая анемия, обусловленная эритроопсонинами. Существование аутоопсонинов к клеткам крови является общепризнанным. При приобретенной идиопатической гемолитической анемии, циррозе печени, гипопластической анемии с гемолитическим компонентом и лейкозах обнаружен феномен аутоэритрофагоцитоза. Приобретенная идиопатическая гемолитическая анемия, сопровождающаяся положительным феноменом аутоэритрофагоцитоза, имеет хроническое течение. Периоды ремиссии, длящиеся иногда значительное время, сменяются гемолитическим кризом, характеризующимся иктеричностью видимых слизистых оболочек, потемнением мочи, анемией, ретикулоцитозом и повышением непрямой фракции билирубина, иногда увеличением селезенки и печени. При идиопатических и симптоматических гемолитических анемиях выявление аутоэритрофагоцитоза при отсутствии данных, указывающих на наличие других форм аутоиммунных гемолитических анемий, дает основание отнести их к гемолитической анемии, обусловленной эритроопсонинами. Диагностическая проба аутоэритрофагоцитоза проводится в прямом и непрямом вариантах. Иммуногемолитические анемии, обусловленные применением лекарств. Различные лечебные препараты (хинин, допегит, сульфаниламиды, тетрациклин, цепорин и др.), способные вызывать гемолиз, образуют комплексы со специфическими гетероантителами, затем оседают на эритроциты и присоединяют к себе комплемент, что приводит к нарушению мембраны эритроцитов. Такой механизм медикаментозно обусловленных гемолитических анемий подтверждается обнаружением на эритроцитах больных комплемента при отсутствии на них иммуноглобулинов. Анемии характеризуются острым началом с признаками внутрисосудистого гемолиза (гемоглобинурия, ретикулоцитоз, повышение содержания свободной фракции билирубина, усиление эритропоэза). На фоне гемолитического криза иногда развивается острая почечная недостаточность. Несколько по-иному протекают гемолитические анемии, развивающиеся при назначении пенициллина и метилдофа. Введение за сутки 15 000 и более ЕД пенициллина может привести к развитию гемолитической анемии, характеризующейся внутриклеточным гипергемолизом. Наряду с общими клинико-лабораторными признаками гемолитического синдрома обнаруживается также положительная прямая проба Кумбса (выявляемые антитела относятся к IgG). Пенициллин, связываясь с антигеном мембраны эритроцитов, образует комплекс, против которого в организме вырабатываются антитела. Гемолитико-уремический синдром (болезнь Мошковича, синдром Гассера) может осложнять течение аутоиммунных гемолитических анемий. Заболевание аутоиммунной природы характеризуется гемолитической анемией, тромбоцитопенией, поражением почек. Отмечаются диссеминированное поражение сосудов и капилляров с вовлечением практически всех органов и систем, выраженные изменения со стороны коагулограммы, характерные для ДВС-синдрома. В остром периоде заболевания наблюдается тромбоцитопенический геморрагический синдром, часто носящий генерализованный характер: от гемморагических высыпаний на коже и слизистых оболочках до кровоизлияний в жизненно важные органы, включая головной мозг. Верификация диагноза Лечение аутоиммунных гемолитических анемий Спленэктомия при аутоиммунной гемолитической анемии, связанных с тепловыми агглютининами и аутоэритроопсонинами, может быть рекомендована лишь больным, у которых кортикостероидная терапия сопровождается непродолжительными ремиссиями (до 6—7 мес.) или имеется резистентность к ней. У больных гемолитической анемией, обусловленной гемолизинами, спленэктомия не предотвращает гемолитические кризы. Однако они наблюдаются реже, чем до операции, и легче купируются с помощью кортикостероидных гормонов. При рефрактерных аутоиммунных гемолитических анемиях в сочетании с преднизолоном могут быть использованы иммунодепрессаты (6-меркаптопурин, имуран, хлорбутин, метотрексат, циклофосфамид и др.). В стадии глубокого гемолитического криза применяют переливания эритроцитной массы, подобранной с помощью непрямой пробы Кумбса; для снижения выраженной эндогенной интоксикации назначают гемодез, полидез и другие дезинтоксикационные средства. Лечение гемолитико-уремического синдрома, который может осложнять течение аутоиммунных гемолитических анемий, включает кортикостероидные гормоны, свежезамороженную плазму, плазмаферез, гемодиализ, трансфузии отмытых или криоконсервированных эритроцитов. Несмотря на использование комплекса современных терапевтических средств, прогноз часто неблагоприятный.
047. Апластическая анемия. Апластическая анемия - состояние, характеризующееся снижением гематопоэтической активности (депрессией) костного мозга с развитием наряду с анемией также лейко- и тромбоцитопении. Этиологии и патогенез. Апластическая анемия может развиться при воздействии ряда миелотоксических факторов: ионизирувщего излучения, химических веществ - бензола, солей золота, мышьяка; лекарственных средств - хлорамфеникола (левомицетина), фенил-бутазона (бутадион), хлорпромазина (аминозин), мепро-бамата, дилантина, антиметаболитов (б-меркаптопурина, метотрексата), алкилирующих (циклофосфана, хлорбути-на) и некоторых других средств. Миелотоксический эффект от воздействия одних факторов (ионизирующее излучение, антиметаболиты) возникает всегда при достаточно большой дозе, других - проявляется индивидуально. Причина индивидуальной чувствительности, в частности к некоторым лекарственным средствам не всегда ясна, но может быть связана с генетическими дефектами кронетнорных клеток. Это относится, например, к хлорамфениколу и фенилбутазону, которые вызывают супрессию (в зависимости от дозы) эритропоэза с частотой соответственно 1:24000 и 1:40000 лиц, их принимающих. Существует и наследственная форма апластической анемии - анемия Фанкони. Более чем у половины больных не удается выявить какие-либо причинные факторы - это так называемая идгопатическая апластическая анемия. Механизмы, лежащие в основе идиопатической формы анемии, неясны. Возможен аутоиммунный механизм, связанный с воздействием на клетки костного мозга аутоантител при участии иммунных лимфоцитон. Показано, что лимфоциты (Т-супрессоры) больных тормозят образование эритроцитных колоний костного мозга донора и могут нарушать дифференциацию и пролиферацию гематопоэтических предшественников. Предполагают также, что основой апластической анемии может быть поражение (внутренний дефект) стволовой клетки, о чем свидетельствует восстановление кроветворения у больных после трансплантации им аллогенного костного мозга, содержащего нормальные стволовые клетки. Существуют экспериментальные данные, свидетельствующие о значении для развития апласткческого процесса и нарушений микроокружения - первичного дефекта стромальных клеток костного мозга. Однако суть этих клеточных дефектов остается неясной, так же как и их первичность. Возможно, что при разных формах апластической анемии патогенетические механизмы неодинаковы. Клиническая картина. Идиопатическая апластическая анемия может начинатьсл остро и иметь быстро прогрессирующее течение, но чаще болезнь развивается постепенно. Клинические проявления зависят от выраженности цитопении. При физическом исследовании отмечаются бледность кожных покровов, одышка, тахикардия, выслушивается систолический шум в области сердца. Геморрагический синдром соответствует степени тромбоцитопении, наблюдаются петехиальные высыпания на коже и слизистых оболочках, кровоточивость десен, носовые и маточные кровотечения. Следствием нейтропении являются инфекционные осложнения - ангины, пневмонии, инфекция мочевых путей, сепсис. Селезенка обычно не увеличена. Л а б о р а т о р н ы е д а н н ы е. Гематологическими признаками аплазии костного мозга являются выраженная анемия (концентрация гемоглобина может падать до 20 - 30 г/л), лейкопения (нейтропения с относительным лимфоцитозом) и тромбоцитопения, иногда до полного исчезновения тромбоцитов из крови. Анемия чаще нормохромная и макроцитарная (СЭО) 94 мкм отмечается примерно у 60 - 65 % больных), число ретикулоцитов снижено. Содержание железа в сыворотке крови нормальное или повышенное, насыщение трансферрина близко к 100 %. В ряде случаев отмечается повышение уровня фетального гемоглсбина (НЬР составляет до 15% от общего гемоглобина) и эритропоэтина (поскольку продукция эритроцитов резко снижена, то либо существует ингибитор эритропоэтина, либо костный мозг к нему нечувствителен). СОЭ обычно увеличена до 40 - 60 мм/ч. При пункционной биопсии костного мозга получают малое количество ядросодержащих клеток (миелокариоцитов) или они совсем отсутствуют, при гистологическом исследоваиии отмечают замещение гемопоэтической ткани жировой тканью, Однако даже если биопсию производят в разных местах, то она не отражает состояние всего костного мозга: на аутопсии часто обнаруживают островки кроветворения (регенерации), содержащие двуядерные и многоядерные эритроидные клетки, среди значительно опустошенного костного мозга. При быстром прогрессировании болезни смерть может наступить через несколько месяцев, при хроническом течении происходит смена обострения и ремиссий. Иногда наблюдается полное выздоровление. Апластическая анемия Фанкони - наследственная, часто семейная аномалия, передающаяся по аутосомно-рецессивному типу; выявляется у детей в возрасте от 4 до 10 лет, характеризуется, помимо аплазии костного мозга и панцитопении, также рядом соматических и метаболических нарушений: задержкой роста, дефектами формирования скелета, микроцефалией, гипогонадизмом, гипоплазией почек, аминоацидурией, глюкозурией, гиперпигментацией кожи. Изменения показателей крови часто менее выражены, чем при идиопатической форме апластической анемии. Наблюдается склонность к развитию острого лейкоза. Диагноз и дифференциальный диагноз. Апластическую анемию предполагают при снижении в крови количества эритроцитов, лейкоцитов и тромбоцитов (панцитопения). Диффереициальную диагностику следует проводить с острым лейкозом и В12-дефицитной анемией. Важное значение имеет стернальная пункция: при обнаружении гиперплазии эритроцитарного ростка апласти-ческая анемия исключается. Если увеличенные в количестве эритроидные элементы имеют морфологические черты мегалобластов, то вероятно наличие у больного В12-дефицитной анемии, но при этом необходимо иметь в виду и возможность эритромиелоза. Присутствие в костномозговом пунктате большого количества бластных клеток свидетельствует об остром лейкозе. Получение при пункции скудного материала еще не может быть расце-нено как указание на аплазию костного мозга, так как такая картина бывает и при остром алейкемическом лейкозе. Для уточнения диагноза производят трепано-биопсию кости; уменьшение активного кроветворного костиого мозга и увеличение жировой ткани подтверждает диагноз апластической анемии. Апластическую анемию необходимо дифференцировать и с иммунной периферической цитопенией. Для диагностики иммунной цитопении имеют значение обнаружение увеличенной селезенки, положительные проба Кумбса или агрегат-гемагглютационная проба, нормальное количество мегакариоцитов в костном мозге. Отграничение апластической анемии от пароксизмальной ночной гемоглобинурии и гемолизиновых форм аутоиммунной гемолитической анемии основывается на отсутствии при апластической анемии признаков внутрисосудистого гемолиза, ретикулоцитоза, увеличения селезенки. Лечение и прогноз. Лечение направлено на коррекцию цитопенического синдрома и костномозговой недостаточности и борьбу с осложнениями. Вначале отменяют все лекарственные средства, к которым у больного имеется индивидуальная повышенная чувствительность и которые могут быть причастны к развитию анемии. При тяжелой анемии проводят заместительные трансфузии отмытых эритроцитов, при выраженной тромбоцитопении и геморрагиях - переливания тромбоцитарной массы (лучше от одного донора). При инфекционных осложнениях применяют антибиотики широкого спектра действия. Наиболее перспективным методом лечения апластиче-ской анемии является трансплантация аллогенного совместимого по НЕА-антигенам костного мозга. Трансплантация костного мозга показана лицам молодого возраста, особенно с тяжелыми, прогностически неблагоприятными формами болезни (уровень тромбоцитов ниже 20 ° 10"/л, нейтрофилов менее 0,5 ° 10"!л, количества ретикулоцитов после коррекции менее чем 1 % и числа клеток костного мозга менее 25 % от общего объема). Трансплантация при наличии подходящего донора должна быть ранней, когда аллоиммунизация вследствие заместительной терапии еще невелика (подробнее о трансплантации). В случаях невозможности трансплантации предпринимают попытку лечения преднизолоном в высоких дозах (60 - 80 мг/сут), при отсутствии эффекта - в небольших дозах, в основном с гемостатической целыо. У ряда больных (по некоторым данным, более чем у половины) оказывается успешной спленэктомия (в связи с удалением органа, вырабатывающего антитела). Спленэктомия показана при менее тяжелых формах болезни - отсутствии большой кровоточивости и признаков сепсиса. Эффект наступает через 2 - 5 мес после операции, но кровоточивость обычно уменьшается сразу. После спленэктомии проводят лечение анаболическими гормонами (неробол по 20 мг/сут, анаполон по 200 мг/сут в течение полугода). Для лечения апластической анемии применяют также антилимфоцитарный глобулин, по данным зарубежных авторов, использование коммерческого антилимфоцитарного глобулина эффективно у 40 - 50% больных, что позволяет рекомендовать его на ранних этапах лечения. По опыту отечественных гематологов антилимфоцитарный глобулин целесообразно назначать после спленэктомии (при ее неэффективности или как подкрепляющую терапию), кроличий и козий антилимфоцитарный глобулин отечественного производства вводят внутривенно по 120 - 160 мг 10 - 15 раз.
048. Агранулоцитоз — это уменьшение числа лейкоцитов (менее 1000 в 1 мкл) или числа гранулоцитов (менее 750 в 1 мкл крови). Агранулоцитоз, как правило, представляет собой синдром какого-то общего заболевания. Классификация Чаще встречается миелотоксический агранулоцитоз (см. Цитостатическая болезнь) и иммунный. Последний может быть обусловлен появлением аутоантител (например, при системной красной волчанке) и антител к гранулоцитам после приема медикаментов, оказавшихся гаптенами (при попадании в организм эти медикаменты, соединяясь с белком, обретают свойства антигена). Гаптеновый агранулоцитоз развивается под влиянием диакарба (диамокса), амидопирина, антипирина, ацетилсалициловой кислоты, барбитуратов, изониазида (тубазида), мепротана (мепробамата), фенацетина, бутадиона, новокаинамида (прокаинамида), индометацина, левамизола, сульфаниламидов, метициллина, триметоприма (входит в состав бактрима), хингамина (хлорохина), инсектицидов, клозапина (лепонекса) и др. Патогенез Изучен недостаточно. При аутоиммунных формах поражения преждевременная гибель гранулоцитов и их костномозговых предшественников обусловлена аутоантителами. Механизм индивидуальной реакции организма на введение медикамента при гаптеновом агранулоцитозе неясен. Однажды появившись, гаптеновый агранулоцитоз будет неизменно повторяться при введении в организм того же препарата — гаптена. Симптомы Клиническая картина болезни обусловлена агранулоцитозом, для которого характерны септические осложнения: ангины, пневмонии и т. п. При гаптеновом агранулоцитозе гранулоцитов в крови обычно нет, но число лимфоцитов, тромбоцитов, ретикулоцитов нормальное. Геморрагии не бывает. При аутоиммунном агранулоцитозе изредка возможно появление антител и к тромбоцитам, тогда возникает тромбо   ЧТО ПРОИСХОДИТ, КОГДА МЫ ССОРИМСЯ Не понимая различий, существующих между мужчинами и женщинами, очень легко довести дело до ссоры...  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования... 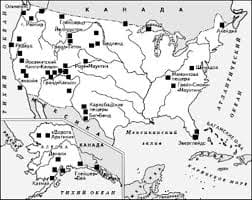 Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  Что будет с Землей, если ось ее сместится на 6666 км? Что будет с Землей? - задался я вопросом... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|