
|
|
Неиммунные тромбоцитопении потребления: тромботическаяТромбоцитопеническая пурпура (ттп) — болезнь мошковиц И гемолитико-уремический синдром (гус) — синдром гассера Инициальным фактором развития ТТП и ГУС являются бактериальная (Shigella dysenteriae, энтеротоксичные штаммы Е. Coli) и вирусная инфекции, иммунизация, оральные контрацептивы, системные заболевания соединительной ткани. ТТП встречается в любом возрасте, ГУС — в детском. Предполагается, что повреждение эндотелия капилляров микробными токсинами и цитокинами приводит к выбросу из эндотелиоцитов большого количества мультимеров фактора Виллебранда, что ведёт к агрегации тромбоцитов и образованию гиалиновых тромбов в сосудах микроциркуляторного русла, к дальнейшим прогрессирующим поражениям стенки сосудов, к развитию гемолиза эритроцитов. В гиалиновых тромбах содержатся, помимо тромбоцитов, нити фибрина, однако генерализованной гиперактивации плазменных факторов системы гемостаза не происходит, что отличает данную патологию от ДВС-синдрома. При ТТП отложение гиалиновых тромбов носит системный характер, а при ГУС поражаются преимущественно микрососуды почек, что обусловливает развитие острой почечной недостаточности. Клиническая картина ТТП и ГУС проявляется: 1. тромбоцитопенией потребления, сопровождающейся кровотечением из носа, желудочно-кишечного тракта (рвота цвета кофейной гущи, кровавый понос), кожной петехиально-экхимозной сыпью; 2. микроангиопатическои гемолитической анемией; 3. лихорадкой; 4. перемежающимися неврологическими нарушениями; 5. почечной недостаточностью (олиго-, анурией, азотемией). При лабораторной диагностике ТТП и ГУС выявляются: ¨ тромбоцитопения (20-40-109/л); ¨ признаки микроангиопатическои гемолитической анемии (снижение содержания в крови эритроцитов и гемоглобина, ретикулоцитоз; в мазке крови — шистоциты); ¨ повышение в крови уровня непрямого билирубина; ¨ увеличение времени кровотечения; ¨ АПТВ, фибриноген, продукты деградации фибрина в норме, что используется при дифференциальной диагностике с ДВС-синдромом; ¨ характерные гиалиновые тромбы в микрососудах при биопсии дёсен (ТТП) или почек (ГУС); ¨ повышенное содержание свободного НЬ плазмы. При тяжёлых формах тромбоцитопении потребления возможен летальный исход. Тромбоцитопатии Тромбоцитопатии бывают – наследственные (первичные), которые развиваются при генных дефектах: (тромбастения Глянцмана, болезнь Виллебранда, болезнь Бернара – Сулье); – приобретенные(симптоматические), вторичные – возникают при: заболеваниях и синдромах (опухоли, ДВС-синдром, пороки сердца, уремия, парапротеинемия, иммунные тромбоцитопении, диффузные заболевания соединительной ткани, заболевания печени и почек, мегалобластные анемии, острые лейкозы, миелопролиферативные заболевания) и при воздействии лекарственных препаратов (аспирин, курантил, нестероидные противовоспалительные средства и др.). Различают следующие основные звенья патогенеза тромбоцитопатий: – нарушение процессов синтеза и накопления в гранулах тромбоцитов биологически активных веществ; – расстройство процессов дегрануляции и высвобождения тромбоцитарных факторов в плазму крови; – нарушение структуры и свойств мембран тромбоцитов. Тромбоцитопатии проявляются: – геморрагическим синдромом – кровотечения носовые, десневые, почечные, желудочно-кишечные, маточные; кровоизлияния в кожу (мелкоточечная геморрагическая сыпь, экхимозы), мышцы, слизистые, подкожную жировую клетчатку, ЦНС, во внутренние органы, полости и др.; – расстройствами микрогемоциркуляции, ведущими к нарушениям обмена веществ в тканях (капилляротрофическая недостаточность); дистрофиями, эрозиями, изъязвлениями; изменениями свойств тромбоцитов (адгезивных, агрегационных, прокоагуляционных); дефектами гранул тромбоцитов – отсутствие или уменьшение их числа (например, при синдроме «серых» тромбоцитов), нарушениями высвобождения их содержимого, отклонением от нормы размера и формы мегакариоцитов и тромбоцитов; – анемическим синдромом – общая слабость, бледность, головокружение, систолический шум на верхушке и др. (при кровотечениях). Тромбастения гланцмана Развитие тромбоцитарной дисфункции обусловливается отсутствием или дефектом мембранного рецептора к фибриногену и гликопротеинам IIb - IIIa. Это приводит к резкому снижению интенсивности процесса связывания фибриногена с мембраной тромбоцита, в результате чего нарушается агрегация тромбоцитов. Заболевание наследуется аутосомно-рецессивно, проявляется уже в раннем детском возрасте, характеризуется петехиально-экхимозным типом кровоточивости, склонностью к кровотечению из слизистых оболочек (носовые, маточные кровотечения, кровоизлияния в склеру и сетчатку глаза), длительными кровотечениями после удаления зуба или ЛОР-операций. При исследовании семейного анамнеза в родословной выявляется про-банд больных родственников в семьях обоих родителей «по горизонтали». При лабораторной диагностике обнаруживаются: - увеличение времени кровотечения; - нормальное количество тромбоцитов; - в пределах нормы адгезия тромбоцитов, при изучении ристоцетин-индуцированной агрегации тромбоцитов выявляется отсутствие типичной двухфазной кривой; - АПТВ в норме. СИНДРОМ (БОЛЕЗНЬ) БЕРНАРА-СУЛЬЕ (МАКРОЦИТАРНАЯ ТРОМБОЦИТОДИСТРОФИЯ, СИНДРОМ ГИГАНТСКИХ ТРОМБОЦИТОВ) При данной болезни в мембране тромбоцита отсутствует специфический гликопротеин, взаимодействующий с ФВ-VIII, ФУ, Ф1Х и ристоцетином, а также повышается содержание сиаловых кислот, снижается электрический заряд. Это приводит к нарушению адгезионных свойств тромбоцитов. Болезнь наследуется аутосомно-рецессивно, характеризуется укорочением продолжительности жизни тромбоцитов при нормальном процессе их продуцирования в костном мозге, следствием чего является развитие умеренной тромбоцитопении. Основным морфологическим критерием заболевания является наличие в крови гигантских тромбоцитов, достигающих в диаметре 6-8 мкм (в норме 2-4 мкм). Клиническая картина характеризуется кровоточивостью петехиального типа, тяжесть которой варьирует в больших пределах — от относительно лёгких и латентных форм до тяжёлых и даже фатальных случаев. Тяжесть кровоточивости зависит от содержания аномальных тромбоцитов: чем выше их процент, тем тяжелее и потенциально опаснее протекает геморрагический синдром. При лабораторной диагностике обнаруживается: - увеличение времени кровотечения; - тромбоцитопения, увеличение размера тромбоцитов; - снижение адгезии тромбоцитов и ристоцетин-индуцированной агрегации; нормальные показатели коагуляционного гемостаза, в том числе АПТВ. Болезнь виллебранда В основе развития заболевания лежит дефицит или функциональная неполноценность фактора Виллебранда (ФВ). Наследуется болезнь аутосомно-доминантно с неполной пенетрантностью или (реже) аутосомно-рецессивно. Дефицит и/или дефект ФВ приводит к нарушению процесса адгезии тромбоцитов к коллагену сосудистой стенки и снижению интенсивности процесса образования комплекса ФВ—ФУТП, а также к уменьшению периода его полусуществования за счёт ускорения катаболизма и элиминации ФУШ из крови. Клиническая картина болезни разнообразна и зависит как от фенотипиче-ского проявления патологического гена, так и от физиологического статуса организма (беременность, стресс, приём контрацептивов и т. д.). Дефицит и/или дефект ФВ приводит к нарушению и сосудисто-тромбоцитарного, и коагуляционного гемостаза. Это проявляется экхимозными, реже — гематомными кровоизлияниями, меноррагиями, кровоточивостью слизистых оболочек. При хирургических вмешательствах характерен высокий риск профузных кровотечений. При лабораторной диагностике выявляются: - увеличение времени кровотечения; нормальное количество тромбоцитов; снижение степени адгезии тромбоцитов к стеклу и ристоцетин-индуцированной агрегации; - снижение содержания и/или активности ФВ; увеличение АПТВ.
  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала... 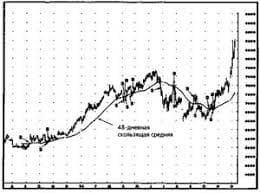 Что вызывает тренды на фондовых и товарных рынках Объяснение теории грузового поезда Первые 17 лет моих рыночных исследований сводились к попыткам вычислить, когда этот...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор...  Что будет с Землей, если ось ее сместится на 6666 км? Что будет с Землей? - задался я вопросом... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|