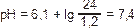|
|
Ы Вёрстка Вставить файл «Формула Приложения Уравнение Хендерсона Хассельбаха 2 Расчет»
• Дыхательная регуляция рСО2 артериальной крови. Лёгкие обладают способностью задерживать или активизировать выделение СО2 и таким образом регулировать кислый компонент бикарбонатной буферной системы. • Почечная регуляция содержания бикарбоната плазмы. Почки при секреции Н+ регулируют содержание бикарбоната плазмы за счёт образования нового бикарбоната. Этот процесс восполняет бикарбонат, используемый для нейтрализации кислот, образующихся при незавершённом метаболизме нейтральных пищевых продуктов и при метаболизме кислых продуктов. Существует два важных аспекта метаболизма Н+ в почках: реабсорбция ионов бикарбоната и секреция Н+. † Реабсорбция ионов бикарбоната. Примерно 4300 мэкв бикарбоната фильтруется ежедневно через клубочки, бикарбонат практически полностью реабсорбируется в проксимальных канальцах почек. Оставшееся минимальное количество бикарбоната реабсорбируется в дистальных канальцах и собирательных трубочках. Проксимальная канальцевая реабсорбция бикарбоната осуществляется косвенно за счёт следующего процесса. ‡ Отфильтрованный бикарбонат и секретируемый H+ в просвете канальца образуют угольную кислоту. Реабсорбция Na+ сопряжена с секрецией H+, что поддерживает состояние электронейтральности. ‡ Карбоангидраза щёточной каёмки клеток проксимальных канальцев катализирует превращение угольной кислоты в двуокись углерода и воду. Двуокись углерода диффундирует в цитоплазму клеток проксимальных канальцев, где внутриклеточная карбоангидраза катализирует её регидратацию в угольную кислоту. ‡ Бикарбонат, образующийся при диссоциации угольной кислоты, пассивно реабсорбируется в кровь вместе с эквимолярным количеством Na+, активно транспортирующегося в кровь. Н+, образующийся при диссоциации угольной кислоты внутриклеточно, служит источником дополнительных Н+, подлежащих секреции. ‡ Вход Na+ в клетку канальца происходит двояко: при пассивной диффузии Cl– внутрь клетки из просвета канальца и при обмене H+ на Na+. Конечный результат диффузии Cl– в клетку и Na+-H+–обмена — реабсорбция NaCl или бикарбоната натрия соответственно. † Добавление нового бикарбоната. В дополнение к сохраняемому бикарбонату почки добавляют к плазме новый бикарбонат посредством секреции Н+. ‡ Добавление нового бикарбоната касается не столько бикарбоната, реабсорбирующегося в проксимальных канальцах, сколько бикарбоната, накапливающегося внутри клеток дистальных канальцев путём гидратации двуокиси углерода и последующей диссоциации угольной кислоты. ‡ Участие почек в образовании нового бикарбоната сопровождается экскрецией эквивалентного количества кислоты в мочу в виде титруемой кислоты, иона аммония или их обоих. ‡ Образование и секреция титруемой кислоты. Обмен Н+ на Na+ превращает двухосновный фосфат или сульфат натрия в фильтрате в одноосновный фосфат или сульфат натрия, экскретирующиеся с мочой в виде титруемой кислоты. Вследствие этого Н+, секретируемый в дистальные канальцы, может реагировать с фосфатом, а не с бикарбонатом. ‡ Образование и секреция аммиака. В отличие от фосфата, аммиак попадает в просвет канальца не за счёт фильтрации, а за счёт образования в канальцах и секреции. Фактически весь аммиак, проникающий в просвет канальцев, немедленно соединяется с Н+ с образованием иона аммония, не обладающего способностью к диффузии вследствие его нерастворимости в жирах. В результате экскреция иона аммония почками приводит к добавлению бикарбоната к плазме. † Концепция компенсации. Компенсацию можно определить как физиологический ответ на изменение или респираторного, или метаболического (почечного) компонента в КЩР для восстановления нормальной величины рН. Физиологическая компенсация обычно бывает неполной. В уравнении Хендерсона–Хассельбальха изменения в числителе (метаболический компонент) связаны с вторичными изменениями в знаменателе (респираторный компонент), что восстанавливает логарифм отношения в пределах 24:1,2 (т.е. 20:1); в результате рН поддерживается в пределах нормы. И наоборот, изменения респираторного компонента сочетаются с компенсаторными изменениями метаболического компонента, что также поддерживает рН в пределах нормы. Нарушения половой дифференцировки Нозологические формы • Истинный гермафродитизм. В половых железах присутствует ткань как семенников, так и яичников. Кариотип: приблизительно в 80% — 46XX, остальные случаи — 46XY или мозаицизм. Этиология неясна. † Обычно выражена значительная вирилизация, вследствие чего большинство истинных гермафродитов воспитываются как мужчины. Могут возникать гинекомастия и циклическая гематурия как результат маточного кровотечения. † Серьёзное подозрение в истинном гермафродитизме должно возникнуть, если у ребёнка гениталии переходного типа, ХХ-кариотип и нормальный уровень 17-гидроксипрогестерона, что исключает недостаточность 21-гидроксилазы. Окончательный диагноз базируется на хирургическом исследовании и обнаружении половых желёз, содержащих ткани как яичника, так и семенников. • Смешанный дисгенез половых желёз наблюдается при кариотипе 45Х/46XY. † Клиника: широкий спектр строения внешних половых органов — от полностью мужских до полностью женских. ‡ Половые железы могут выглядеть различно: от узнаваемых внешне яичников до дисгенетических яичек; часто наблюдают асимметричность гонад. ‡ Эффекты клеточной линии 45Х могут имитировать фенотип синдрома Шерешевского – Тёрнера. † Диагноз устанавливают кариотипированием. • Мужской псевдогермафродитизм. Дети генотипа 46,XY; имеются яички, но маскулинизация неполная (гипоспадия, микрофаллия, недоразвитая мошонка с яичками или без них). Мужской псевдогермафродитизм наблюдают при множестве эндокринных расстройств (дефекты синтеза тестостерона, его метаболизма и эффектов на клетки-мишени). † Райфенштайна синдром. Семейная форма мужского псевдогермафродитизма: фенотипически неопределённая половая принадлежность гениталий, гипоспадия, постпубертатная гинекомастия; бесплодие в связи со склерозом семенных канальцев. Синоним: Кляйнфелтера – Райфенштайна – Олбрайта синдром. † Тестикулярная феминизация (см. «Синдром тестикулярной феминизации »). † Псевдогермафродитизм мужской (*300018, Xp21.3, ген DSS, • Недостаточность 5 • Нарушения синтеза и метаболизма тестостерона наблюдают редко; известно несколько форм ферментной недостаточности ( • Женский псевдогермафродитизм. Дети генотипа 46,XX (имеются яичники), но, как правило, мужской фенотип при рождении. Повышенная чувствительность ХХ-плода к воздействию андрогенов во время критического периода (8 нед внутриутробного развития) приводит к развитию разной степени выраженности губо-мошоночного сращения, формированию урогенитального синуса и увеличению клитора. Некоторые дети при рождении выглядят как мальчики с крипторхизмом. † Врождённая гиперплазия надпочечников. Дефекты, приводящие к развитию женского псевдогермафродитизма, — недостаточность 21- и 11-гидроксилазы, а также 3 • Врождённые пороки наружных половых органов: гипоспадия и микрофаллия. † Гипоспадию различной степени выраженности регистрируют изолированно или в сочетании с другими врождёнными дефектами, особенно мочеполовой системы. † Микрофаллия. Гениталии у мальчиков небольших размеров, но хорошо дифференцированные. Существуют стандарты оценки длины выпрямленного полового члена, начиная с раннего детского возраста до взрослого состояния. Рост гениталий определяют гормональной стимуляцией яичек плода гипофизарным гонадотропином. ‡ Этиология. Микрофаллия (гипоплазия полового члена) может указывать на постнатальный гипогонадотропный гипогонадизм (как при синдроме Калльмана) или может быть отражением врождённого гипопитуитаризма. ‡ Лечение. Можно ожидать эффекта от применения тестостерона (25–50 мг каждые 3 нед в течение 3 мес), что приводит к положительному косметическому эффекту без значительного ускорения созревания скелета. Ведение ребёнка с бисексуальными гениталиями. • Полное диагностическое обследование должно быть предпринято в возможно более ранние сроки после рождения ребёнка с бисексуальными гениталиями. Необходимо убедить родителей отложить присвоение ребёнку имени и атрибуцию пола до окончания диагностических мероприятий. Нужны: тщательный сбор семейного анамнеза, детали течения беременности, общий осмотр. † Осмотр. Следует оценить размеры полового члена, расположение мочеиспускательного канала, наличие пальпируемых гонад (обычно яичек) и другие признаки (дисморфические и асимметричные). † Лабораторные исследования включают: хромосомный анализ, определение содержания электролитов, тестостерона, ЛГ, ФСГ и 17-гидроксипрогестерона. Радиографическое контрастное исследование урогенитальных синусов часто помогает обнаружить влагалище и шейку матки, иногда удаётся рассмотреть маточные трубы. УЗИ органов таза может выявить наличие яичников и матки. ‡ При кариотипе 46,XX и повышенном содержании 17-гидроксипрогестерона наиболее вероятна недостаточность 21-гидроксилазы. При нормальном уровне 17-гидроксипрогестерона наиболее вероятен истинный гермафродитизм. Оценка содержания 11‑дезоксикортизола и дегидроэпиандростерона исключает возможность недостаточности 11-гидроксилазы или 3 • Присвоение пола. Для решения множества терапевтических и общих вопросов необходим точный диагноз. † Даже заметно вирилизированные девочки с недостаточностью 21‑гидроксилазы должны воспитываться как девочки, поскольку при адекватном лечении заболевания и косметическом восстановлении наружных половых органов они будут иметь полноценные репродуктивные возможности. † Для мальчиков с кариотипом 46,XY и бисексуальными гениталиями определение пола необходимо основывать на решении возможности выполнения половых функций в качестве мужчины. Обычно это зависит от размера полового члена и оценки хирургом возможности оперативной коррекции гипоспадии. ‡ Дисгенетические яички и яичнико-семенники должны быть удалены, поскольку высока вероятность их злокачественной трансформации. ‡ Во избежание нежелательных гормональных влияний в период полового созревания необходимо удалять гонады, не соответствующие полу. Лечение. • Адекватная заместительная гормональная терапия, как правило, должна назначаться в период полового созревания. • Генетическое консультирование (в том числе по вопросам полового поведения) семей и детей — важный момент медицинской помощи. • Полное, но тактичное информирование о результатах всех анализов (в соответствии с возрастом пациента) должно привести к успешной психосексуальной адаптации. • Удаление гонад. Вследствие возможной малигнизации ХY‑гонаду при синдроме резистентных яичников необходимо удалить до полового созревания или сразу после диагностики. Нет необходимости удаления гонад у больных ни при синдроме Тёрнера, ни при синдроме Калльмана, поскольку нет потенциальной возможности малигнизации гонад; при синдроме Тёрнера хромосомыY нет, а при синдроме Калльмана хромосомы нормальные. Для синдрома нечувствительности к андрогенам (синдром тестикулярной феминизации) характерно наличие гонад типа ХY; гонады не надо удалять до завершения полового развития, поскольку риск развития новообразований гонад низок до 20-летнего возраста. • Изменение пола производят при гермафродитизме, а также по желанию транссексуала (600952,?), когда больной убеждён, что имеющиеся у него половые признаки не соответствуют его полу. После тщательной предоперационной подготовки (консультация психиатра, заместительная гормональная терапия) выполняют разрушающую операцию с последующей реконструкцией органов при помощи различных лоскутов и кожных трансплантатов. См. также «Бесплодие», «Синдром Кляйнфелтера», «Синдром тестикулярной феминизации», «Дефекты рецептора андрогенов». Нёбо готическое — высокое и узкое н. (микросимптом синдрома Марфана). Невус — пигментированное образование нейроэктодермального происхождения на коже, в состав которого входят невусные клетки, содержащие меланин. Недостаточность
Антитромбина III н. ( Аортальная н. — неспособность клапана аорты эффективно препятствовать обратному движению крови из аорты в левый желудочек во время диастолы желудочков сердца, обусловленная неполным смыканием или перфорацией полулунных заслонок. Белка (протеина) С н. (*176860, 2q13‑q14, дефект гена PROC, Белка (протеина) S н. (*176880, 3p11.1‑q11.2, дефект гена PROS1, Клапана лёгочного ствола н. — неспособность клапана лёгочного ствола эффективно препятствовать обратному движению крови из лёгочного ствола в правый желудочек во время диастолы желудочков сердца, связанная с неполным смыканием или перфорацией полулунных заслонок. Клапана трёхстворчатого н. — неспособность правого предсердно‑желудочкового клапана эффективно препятствовать обратному движению крови из правого желудочка в правое предсердие во время систолы желудочков сердца, связанная с неполным смыканием или перфорацией створок клапана. Митральная н. — неспособность левого предсердно‑желудочкового клапана эффективно препятствовать обратному движению крови из левого желудочка в левое предсердие во время систолы желудочков сердца, связанная с неполным смыканием или перфорацией створок клапана. Недостаточность ферментов (н.ф.). Синдромы врождённых нарушений обмена веществ (например, фенилкетонурия, гомоцистинурия, гликогенозы и сотни других) встречаются редко, но оказывают значительное влияние на физическое, интеллектуальное, психическое развитие и качество жизни. LCAT н., см. «Н. лецитин-холестерин ацилтрансферазы». Альдолазы н. Фруктозо-1,6-дифосфат а. (триозофосфат лиаза, КФ 4.1.2.13, 3 изофермента: аа. 1, 2 и 3, или А, B и C) — гликолитический фермент, катализирующий обратимое превращение фруктозо-1,6-дифосфата в глицеральдегид 3-фосфат и дигидроксиацетон фосфат. А.A экспрессируется в тканях плода и в скелетных мышцах (5% от всего мышечного белка), а.В — в печени, почках, кишечнике, а.A и а.C — в нервной ткани. Известны наследственные заболевания, развивающиеся вследствие недостаточности разных аа. А. А н. (103850, 16q22–q24, ген ALDOА, А. В н. (229600, 9q22, ген ALDOВ, Аденилат киназы н. Фермент (*103000, КФ 2.7.4.3, 9q34.1, известно 3 фенотипа) экспрессируется в эритроцитах и скелетных мышцах. При н. эритроцитарной формы (ген АК1, Аденилосукцинат лиазы н. Для н.ф. (*103050, аденилосукциназа, КФ 4.3.2.2, 22q13.1, ген ADSL, Аденин фосфорибозилтрансферазы н. (*102600, КФ 2.4.2.7, 16q24, ген APRT, не менее десятка дефектных аллелей [носительство около 0,1%, но значительно выше у японцев], Аденозилгомоцистеиназы н. Фермент (*180960, S-аденозил-L-гомоцистеин гидролаза, КФ 3.3.1.1, 20cen–q13.1, ген SAHH, Аденозин монофосфат дезаминазы н. Фермент (адениловой кислоты дезаминаза, аденозиндезаминаза, КФ 3.5.4.6, гены AMPD, Аденозинтрифосфатазы н. (*102800, АТФаза, КФ 3.6.1.3, Аконитазы н. (*255125, Активатора плазминогена н. (*173370, КФ 3.4.21.68, 8p12–8p11, ген PLAT) — сериновая протеаза, активирующая превращение плазминогена в плазмин. При н. ( Аланин - глиоксилат аминотрансферазы н., см. «Оксалоз». Алкилдигидроксиацетонфосфат - КоА синтетазы жирных кислот н., развивается точечная хондродисплазия. Альдостерон синтетазы н., см. «Н. кортикостерон метил оксидазы». Аминоадипиновой семиальдегид синтетазы н., см. «Гиперлизинемия». Аминоацил - гистидин дипептидазы н., см. «Н. карнозиназы».
Аргиназа, см. «Н. ферментов цикла мочевины». Аргининосукцинат лиазы н., см. «Н. ферментов цикла мочевины». Аргининосукцинат синтетаза, см. «Н. ферментов цикла мочевины». Арилсульфатазы н. (КФ 3.1.6.1 [сульфатаза, арил-сульфат сульфогидролаза]) приводит к развитию метахроматической лейкодистрофии (а.А, КФ 3.1.6.8, 250100), мукополисахаридоза (а.В, КФ 3.1.6.12, 253200), ихтиоза (а.С, КФ 3.1.6.2, 308100), точечной хондродисплазии (а.E, 302950). Ароматаза — фермент (*107910, эстроген синтетаза, КФ 1.14.14.1, 15q21.1, ген CYP19) из семейства цитохромов P450 — в яичниках, плаценте, мышцах, печени, жировой ткани, мозге, волосяных фолликулах катализирует образование ароматических С18 эстрогенов из С19 андрогенов (ароматизация). В плаценте активность а. критична для защиты женского плода от маскулинизации и матери от вирилизации. Н.а. ( N- Ацетил - N- Ацетилглутамат синтетаза (КФ 2.3.1.1, ген NAGS). При недостаточности фермента (*237310) развивается гипераммониемия. N- Ацетилглюкозамин -6- сульфатазы н., см. «Мукополисахаридозы». N- Ацетилглюкозаминилфосфотрансферазы н. (КФ 2.7.8.17, 4q21–4q23, ген GNPTA), см. «Муколипидоз». Ацетилгидролаза фактора активации тромбоцитов PAF (*601690, 6p21.1–p12, ген PAFAH2). При н. фермента ( Ацетил - КоА ацетилтрансфераза (КФ 2.3.1.9, ацетил-КоА C-ацетилтрансфераза, ацетоацетил-КоА тиолаза). Известно две формы фермента. • Ацетил-КоА а.1 (*203750, 11q22.3–q23.1, ген ACAT1, 8 мутантных аллелей, • Ацетил-КоА а.2 (*100678, 6q25.3–q26, ген ACAT2, Ацетил - КоА: Ацил - КоА дегидрогеназ н., см. «Дефекты окисления жирных кислот». Ацил - КоА оксидаза, при н. развивается адренолейкодистрофия.
GM1 - Галактокиназы н., см. «Галактоземия». Галактозо - 1 - фосфат уридилтрансферазы н., см. «Галактоземия». Галактозоэпимеразы н., см. «Галактоземия». ГАМК трансаминазы н. (*137150, Гексозаминидазы н. (*268800, Гексокиназы н. Фермент (КФ 2.7.1.1) катализирует реакцию: АТФ + D-гексоза = АДФ + D-гексозо-6-фосфат. Различают 5 изоформ фермента: 1 (*142600, специфичная для эритроцитов, локус 10q22, ген HK1); 2 (601125, мышечная, хр. 2); 3 (142570, г. лейкоцитов, 5q35.2), 4 (138079, глюкокиназа, 7p15, ген GCK), 5 (142550, г. сперматогенных клеток). При н.г.1 (#235700, Гепаран - Гепаран N - сульфатаза, см. « Мукополисахаридозы». Гиалуронидазы н., см. «Мукополисахаридозы». 3- Гидроксиацил - КоА дегидрогеназы н. (бифункциональный фермент, 3-Гидроксиацил-КоА дегидрогеназы (длинноцепочечной) н. (LCHAD, ДКАД) — наследственная ( Биохимия и генетика. • ДКАД (трифункциональный белок митохондрий, локус 2p23) катализирует в митохондриях Проявления заболевания многообразны: синдром внезапной смерти младенца, патология печени (вплоть до фульминантного некроза), кардиомиопатия, миопатия, эпизоды миоглобинурии и острой гипогликемии. Сокращения. ДКАД — длинноцепочечная гидроксиацил-КоА дегидрогеназа (от LCHAD — L ong C hain 3- H ydroxyl-Co AD ehydrogenase).
11 1 11 17 21- Гидроксилазы н., см. «Синдром адреногенитальный», «Гиперплазия коры надпочечника врождённая», «Нарушения половой дифференцировки». 3- Гидрокси - 3 - метилглутарил - КоA лиазы н., см. «Дефекты катаболизма лейцина». 3 11 17 18 20 4- Гидроксифенилпируват гидроксилазы н., см. «Тирозинемия». 25- Гидроксихолекальциферол 1 Гипоксантин гуанин фосфорибозилтрансферазы н. Полная н. фермента (КФ 2.4.2.8) приводит к развитию синдрома Леша – Найена При частичной н. развиваются острый п   Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|