
|
|
Аномалии половых хромосом (гоносом). ⇐ ПредыдущаяСтр 7 из 7 Наряду с хромосомными болезнями, вызванными аномалиями аутосом (хромосомы 1-22), куда входят четко клинически очерченные синдромы с определенными фенотипическими проявлениями и менее специфические с клинической точки зрения хромосомопатии, существует значительная группа синдромов, причинами которых являются нарушения в системе половых хромосом (гоносом). Эти синдромы довольно хорошо изучены, им посвящен целый ряд монографий и большое число публикаций. Отмечено, что грубые пороки ЦНС при гоносомных синдромах встречаются крайне редко, но умственная отсталость при них наблюдается довольно часто (Barnes et al., 1988). В основном, данные синдромы связаны с численными нарушениями хромосом X и У (полными моносомиями, трисомиями, дисомиями), а также структурными изменениями половых хромосом (без участия аутосом) (Wyss et al., 1982; Zuffardi et al., 1997), в том числе и изохромосомами. При этом клинически наблюдают признаки, характерные для известных гоносомных синдромов — синдромы Шерешевского-Тернера, Клайнфельтера, трисомии X, дисомии У. Особо следует отметить ломкую (фрагильную) хромосому X, при которой наблюдают часто встречаемый в популяции синдром умственной отсталости. ХРОМОСОМАХ. Хромосома X — половая субметацентрическая хромосома, исходя из своей средней величины в кариотипе, записанная в группу С. Две хромосомы X в кариотипе человека определяют женский пол: кариотип 46,ХХ. Известна моносомия хромосомы X и полисомии X у девочек, одной из хорошо изученных форм которых является трипло-Х или трисомия X. Моносомия X (синдром Шерешевского-Тернера). Впервые синдром полового инфантилизма, низкорослости и широкой складки на шее описал Н.А..Шерешевский в 1925 г. В 1930 г. Ульрих, проанализировав данные о своих больных с этим симптомокомплексом и литературы, пришел к выводу о том, что речь идет об одном заболевании. После того, как К.Бонневи в 1932 г. описала подобные аномалии у мышей, синдром получил название синдрома Бонневи-Ульриха или Ульриха. И только в 1938 г., когда Х.Тернер выделил у этих больных основную триаду признаков, при этом подчеркнув резкое нарушении функции гонад, синдром во всем мире стали называть синдромом Тернера. В России принято его называть синдромом Шерешевского-Тернера (Шерешевский, 1925; Shereshevsky, 1925; Ulrih, 1930; Turner, 1938). Иногда в мировой литературе до настоящего времени можно встретить его описание как синдрома Ульриха. В дальнейшем клиническую картину синдрома связали с кариотипом 45,Х. Синдром моносомии хромосомы X у девочек, или синдром Шерешевского-Тернера, наиболее изученное хромосомное заболевание, ему посвящены многочисленные исследования. Данные по популяционной частоте синдрома весьма противоречивы. Так, Е.Ф. Давиденкова с соавт. (Давиденкова и др., 1973) оценивает ее как 1 на 3000 девочек, по другим данным — 1 на 6000-7000, причем, мозаичные формы рассматриваются отдельно (Лазюк, 1991). По последним данным, частота синдрома колеблется от 0,1 до 0,4 на 1000. Цитогенетические формы. Есть предположения о том, что все живорожденные дети страдают различными в количественном соотношении (нормальный: аномальный клон клеток) мозаичными формами заболевания, а полные формы элиминируются внутриутробно, т.к. часто в материалах спонтанных абортусов первого триместра беременности обнаруживают кариотип 45,Х во всех клетках. Несомненно, что подтверждение этих предположений нуждается в тщательных молекулярно-цитогенети-ческих исследованиях клеток больных, страдающих синдромом Шерешевского-Тернера. Кроме того, при структурных аномалиях хромосомы X (изохромосомы, делеции короткого и длинного плеча, кольцевые хромосомы, различные Х/Х транслокации) наблюдается клиническая картина синдрома Шерешевского-Тернера. Идентичная клиническая картина при численных и структурных аномалиях хромосомы X можно объяснить феноменом ее инактивации в клеточном цикле, цитогенетическое проявление которой наблюдается в интерфазном ядре в виде хроматина X. Показано, что инактивация одной отцовской или материнской хромосомы X происходит случайно в норме, а в случаях аномальной хромосомы, когда одна из них структурно изменена, обычно инактивируется аберрантная хромосома. Таким образом, девочки со структурными и численными аномалиями хромосомы X клинически неотличимы друг от друга. Клинические проявления. Диагноз синдрома Тернера трудно поставить в роддоме, но уже после первого года жизни ребенка отмечаются такие фенотипические признаки, как антимонголоидный разрез глазных щелей, эпикант, птоз, высокий и широкий лоб, ретрогения, деформированные низко расположенные ушные раковины, короткая шея с крыловидной складкой, низкий рост волос на шее, широкая грудная клетка, вальгусное положение локтей, клинодактилия мизинцев. Из пороков внутренних органов часто отмечают аномалии сердца, сосудов (коарктация аорты) и почек (подковообразная почка, гипоплазия, пиелоэктазия, гидронефроз). Гонады больных представлены соединительнотканными тяжами, в которых находятся недифференцированные клетки или рудименты женских гонад без ова-риальных элементов: отсутствуют также примордиальные фолликулы. Дети рождаются в срок с пренатальной гипоплазией (масса тела до 2500 г) и ростом до 45 см. Однако с возрастом у детей начинает выявляться резко выраженная низкорослость, особенно в препубер-татном периоде, когда этот признак является основным поводом обращения к врачу. В дальнейшем вторичные половые признаки плохо развиты: железистая ткань молочных желез отсутствует, соски недоразвиты, отсутствует оволосение лобка, первичная аменорея (иногда при мозаичных случаях наблюдаются нерегулярные менструации). Интеллект больных приближается к норме. Однако характерным является недоразвитие эмоционально-волевых проявлений: подчиняемость, узость интересов, бедность абстракций, незначительная продуктивность мышления. При выраженных мозаичных формах наблюдают стертую клиническую картину синдрома: отмечают развитие вторичных половых признаков, регулярные менструации. Для таких больных показано гормональное симптоматическое лечение. В допубертатном периоде назначают анаболические стероиды, которые ускоряют рост, в пубертатномпериоде — эстрогены, затем — гестагены. Такая терапия способствует появлению вторичных половых признаков, но больные этим заболеванием остаются бесплодными.
Дисомия Х при мужском кариотипе (синдром Клайнфельтера). Отдельно следует выделить дисомию X при мужском фенотипе как наиболее частую форму гоносомной патологии у мужчин—синдром Клайнфельтера (Klinefelter et al., 1942). Частота встречаемости этого синдрома в популяции 1,2 случая на 1000 новорожденных. Это одна из форм первичного мужского гипогонадизма, в основном, с кариотипом 47.ХХУ, при котором можно наблюдать умственную отсталость с рядом психопатологических признаков. Как правило, новорожденные с этим синдромом не отличаются от своих сверстников: иногда выявляется задержка психомоторного развития и гипоплазия яичек. Основные клинические признаки появляются в препу-бертатном и пубертатном возрасте. Характерным признаком для больных является евнухоидное телосложение с длинными конечностями, узкими плечами и широким тазом. Наружные половые органы сформированы по мужскому типу, но гипоплазированы, резко уменьшен размер яичек, и именно микроорхизм считается одним из важных клинических критериев в диагностике синдрома. Другими диагностическими критериями являются олигофрения, обычно в степени легкой дебильности, и гинекомастия. Однако считать олигофрению постоянным признаком нельзя. Отмечают также аутизм, мнительность, склонность к алкоголизму, асоциальное поведение. Из других признаков известны катаракта, деформация ушных раковин, прогнатизм, алопеция, поперечная ладонная складка, сколиоз, радио-ульнарный синостоз, неврологические нарушения, пороки сердца (Козлова и др., 1996).Однако все эти признаки не имеют диагностического значения. Дальнейшее увеличение числа хромосомХ в кариотипе ведет, как правило, к большей задержке умственного развития и более широкому спектру пороков и микроаномалий развития. Так больные с кариотипом 49,ХХХХУ страдают глубокой олигофренией: их IQ не достигает и 40 единиц, тогда как IQ с классической формой синдрома (кариотип 47.ХХУ) может достигать в отдельных случаях и 80. Существует вариант синдрома Клайнфельтера с кариотипом 48ДХУУ, т.е. с увеличением числа хромосомы У, последняя обеспечивает высокорослость таким больным (Parker et al.,1970). Все возможные кариотипы синдрома Клайнфельтера представлены в таблице, среди которых и 49,ХХХХУ (Singh, Rajkova, 1986).
Полисомии X. В комплексное название синдрома полисомии X входят трисомия (XXX), тетрасомия Х(ХХХХ) и пентасомия Х(ХХХХХ). Несмотря на то, что каждая из этих форм имеет статус синдрома, диагноз больным ставится только после проведения цитогенетического исследования. Это, вероятно, связано с тем, что лишние хромосомы X в кариотипе инактивируются и не проявляют резко выраженного патологического эффекта. Трисомия X. Трисомия X — одна из форм полисомии X, часто встречающаяся и довольно хорошо изученная (Villanueva, Rebar, 1983; Spear, Porto, 1988), особенно с точки зрения психоневрологического статуса (Давиденкова и др., 1973). Самым частым симптомом трисо-мии X является умственная отсталость в стадии дебильности. Следует отметить, что отбор для цитогенетического исследования идет из психиатрических больниц у взрослых индивидуумов и домов инвалидов у детей, которые страдают умственной отсталостью. Несмотря на существование корреляции трисомии X с умственной отсталостью, показано, что дополнительная хромосома X с возрастом увеличивает в два раза риск заболевания каким-либо психозом. Часто у взрослых женщин наблюдают шизофрению с неблагоприятным типом течения и выраженными изменениями личности (Филиппов, 1971). При цитогенетическом обследовании больных шизофренией частота встречаемости трисомии X выше таковой у женщин в общей популяции. Нарушение хромосомного баланса при лишней хромосоме X ведет и к проявлению эпилепсии как в детском, так и в более старшем возрасте. Отмечено также, что темп психической деятельности у таких больных заторможен, двигательная активность снижена, наблюдают также истеричность, различные нарушения поведения с патологией влечений. Соматические аномалии выражены слабо. У большинства девочек они отсутствуют, но иногда можно наблюдать микроцефалию, эпикант, гипертелоризм, косоглазие, уплощение переносицы, высокое небо, неправильное положение зубов, укорочение и искривление мизинцев, кифосколиоз, высокий рост. Добавочная хромосома X приводит к эндокринному дисбалансу, что может вызывать нарушение воспроизводительной функции в дальнейшем (Spear, Porto, 1988). Если функции яичников нормальны, то в будущем эти девочки способны к деторождению. Мозаичные формы для трисомии X не характерны. В целом специальными проявлениями синдрома являются небольшая задержка умственного развития, нарушение поведения и склонность к эпилепсии. Тетрасомия X. При тетрасомии X (кариотип 48.ХХХХ) наблюдается значительная умственная отсталость, отклонения в поведении, эмоциональная неустойчивость, эпилептические припадки. Для таких больных характерны общая гипокинезия, снижение активности, неловкость в движениях. Эпикант, гипертелоризм, близорукость, клинодактилия, высокорослость, нарушения полового развития, наблюдаемые при трисомии X, у больных с тетрасомиями X встречаются наиболее часто (Collen et al., 1985). ПентасомияХ. Следует отметить, что с накоплением хромосом X в кариотипе появляются более выраженные пороки развития (Fragosa et al., 1982). Случаи пентасомии X (кариотип 49.ХХХХХ) встречаются крайне редко. Ведущим признаком заболевания является резко выраженная психомоторная отсталость. На фоне глубокой олигофрении наблюдают монголоидный разрез глазных щелей, эпикант, гипертелоризм, косоглазие, уплощение затылка, короткую шею с низким ростом волос, клинодактилию мизинцев, поперечные складки на ладонях, микромелию, синостоз лучевой и локтевой костей. Из аномалий внутренних органов чаще всего наблюдают врожденные пороки сердца (незаращение артериального протока). У больных в пубертатном и постпубертатном периоде встречают недоразвитие вторичных половых признаков, аменорею. В общем, можно отметить, что максимальное проявление полисомии X характеризуется тяжелыми поражениями интеллекта и выраженными соматическими проявлениями. Ломкая хромосома X, синдром Мартина-Белл. Одной из самых частых форм умственной отсталости у детей является умственная отсталость, сцепленная с ломкой хромосомой X, или синдром FRAX Частота встречаемости этого синдрома в популяции составляет 1 на 1250-1500 среди индивидуумов мужского пола и 1 на 2000-2500—женского. В последних исследованиях появляются данные с несколько иной частотой синдрома. Так, в работах 1996 г. указано на частоту умственной отсталости ассоциированной с ломкой хромосомой X, как 1 на 4000 среди лиц мужского пола и 1 на 6000 — женского (Turner et al., 1996). Возможно, подобные уточнения являются результатом интенсивного молекулярного исследования данной патологии и выделения нескольких форм с разной локализацией гена (например, FRAXA, FRAXE и т.д.). Связь специфической клинической картины синдрома FRAXA с ломким участком длинного плеча хромосомы X заставляет относить это заболевание до настоящего времени к числу хромосомных. Природа ломкости (или фрагильности) хромосомы X была тщательно изучена. Показано, что при цитогенетическом исследовании с дифференциальным окрашиванием хромосом по длине неповрежденная хромосома X имеет в районе ломкого участка темный сегмент, а в случае повреждения этот сегмент приобретает рыхлую структуру, и район ломкости выглядит размытым. При дифференциальном окрашивании прометафазных хромосом картина сегментации позволяет идентифицировать ломкий участок как Xq27.3. Экспрессия ломкости у больных колеблется в широких пределах (от 2 до 68%). Для повышения эффективности и достоверности цитогенетической диагностики предложено использование молекулярно-цитогенетического метода, связанного с ДНК-маркированием хромосомы X в кариотипе в целях точной идентификации ломких хромосом при анализе кариотипа больных. По мнению ряда авторов, ломкая хромосома X предрасполагает к мейотическому нерасхождению хромосом, и поэтому синдром умственной отсталости, сцепленный с ломкой хромосомой X, или синдром FRAX, нередко сопровождается различными формами анеуплоидий. Так, описано сочетание этого синдрома с мозаичными и полными формами синдрома Клайнфельтера, трисомией X, мозаичными формами синдрома Тернера, синдромом Дауна. Синдром FRAX привлекает внимание исследователей не только благодаря высокой частоте в популяции и цитогенетическому 90 феномену фрагильности, но и многочисленными фактами, не соответствующими классическим представлениям о сцепленном с полом наследовании. В середине 80-х годов ученые, проведя комплексный анализ более 200 родословных (96 и 110) семей, отягощенных синдромом FRAX, пришли к выводу о неменделевском характере наследования. Оказалось, что наряду с абсолютным увеличением лиц с признаками заболевания в каждом последующем поколении родословной обнаруживается высокая частота проявления умственной отсталости среди гетерозиготных носительниц. Эти особенности сегрегации синдрома FRAX в родословных получили название «парадокса Шермана» (Sherman et al., 1985). На основании проведенного анализа авторы предположили двухэтапный механизм мутации — от нормального состояния через премутацию к полной мутации. Было выдвинуто, а затем подтверждено предположение о том, что CGG-повторы являются компонентом ломкого сайта хромосомы X и могут быть участком, за счет которого идет амплификация ДНК при мутации в гене. Эти тринуклеотидные повторы расположены в некодирующей части гена, у нормальных индивидуумов представлены 6-50 копиями. В последнее время доказано, что синдром представляет собой пример динамической мутации. У больных с синдромом ломкой хромосомы- Х число копий повтора составляет 230 и более. Такое число называют полной мутацией. Полная мутация возникает не сразу. Сначала появляется «премутация» с числом повторв от 50 до 230. Эта премутация может стать полной мутацией только во время созревания женских половых клеток, но не мужских. Поэтому мужчины с премутацией могут передать ее только своим дочерям. Мужчины и женщины с премутацией клинически здоровы. У женщин, носительниц премутации, во время мейоза могут происходить неожиданное увеличение числа повторов и превращение премутации в полную мутацию. Несмотря на прогресс в молекулярном изучении данного синдрома, цитогенетическое определение ломкой хромосомы X по-прежнему играет важную роль в диагностике заболевания. Сочетание цитогенетического анализа с молекулярными исследованиями позволяет улучшить диагностику сложных и упростить идентификацию уникальных случаев синдрома умственной отсталости, сцепленной с ломкой хромосомой - X. Клинические проявления. Клинически этот синдром характеризуется следующими признаками: макро- и долихоцефалия, удлиненное лицо, широкий прямой лоб с высокой границей волос и увеличенными лобными буграми, гипертелоризм, реже страбизм, уплощенная и гипоплазированная средняя часть лица, высокие аркообразные брови, клювовидный нос, большой выступающий подбородок, толстые губы, увеличенное расстояние между зубами, высокое арковидное небо. Особенно характерным признаком являются оттопыренные низко расположенные ушные раковины, плохо сформированные со сглаженным завитком и противозавитком, а также мягкими мочками. Наиболее важным в диагностическом отношении симптомом синдрома считают увеличение тестикул или макроорхизм, а нередко тотальное увеличение гонад — макрогенитализм с гиперпигментацией мошонки. Нередко отмечают крипторхизм и отсутствие складчатости мошонки. Но самым основным признаком синдрома FRAX считается умственная отсталость. Большинство пациентов страдают умственной отсталостью в степени дебильности. Однако существует широкая вариабельность коэффициента интеллектуальности (IQ) у больных в пределах от 13 до 75 единиц. Показано незначительное снижение интеллекта с возрастом. У детей наблюдается сочетание аутизма с гиперактивностью, двигательной расторможенностью, неадекватным поведением. Предполагают, что проявления аутизма в поведении связаны со структурными изменениями мозжечка и дефектами лобных долей головного мозга (Hagerman, 1990). При исследовании головного мозга с помощью метода ядерно-магнитного резонанса у больных и гетерозиготных носительниц обнаруживают уменьшение размера червя мозжечка, особенно его задней доли, и варолиева моста. Размеры IV желудочка при этом увеличены. Наиболее частыми неврологическими признаками бывают мышечная гипотония, генерализованная гиперрефлексия, спастическая параплегия, координаторные нарушения, неонатальные судороги и единичные эпи-лептиформные приступы, чаще эпизодические на фоне фебрильных состояний, не требующие длительной специфической терапии и напоминающие доброкачественную возрастзависимую эпилепсию (Hecht, 1991). ХРОМОСОМА У. Хромосома У — одна из маленьких по величине хромосом в кариотипе человека. Величина этой половой хромосомы варьирует в зависимости от размера гетерохроматинового района ее длинного плеча и сравнима по длине с хромосомами группы G или между хромосомой 18 и 21-22. Данная хромосома довольно хорошо изучена и картирована (Affara et al., 1996). Эта гоносома определяет мужской пол: кариотип 46,ХУ. Кариотипы без хромосомы X с одной хромосомой У не совместимы с жизнью плода. Отсутствие хромосомы У в кариотипе при мужском фенотипе встречается, в основном, в виде мозаичных форм с хромосомным набором 45,Х/46,ХУ, что ведет к нарушению полового развития у мужчин, у детей же наблюдают малый рост и множественные соматические аномалии (крыловидная складка и низкий уровень волос на шее, антимонголоидный разрез глазных щелей, эпикант, птоз, деформированные низко расположенные ушные раковины, деформация грудной клетки и грудины). В общем, наблюдаемые признаки аналогичны таковым при синдроме Шерешевского-Тернера у девочек. Кроме того, хромосома У характеризуется большим гетерохроматиновым участком длинного плеча, не содержащим уникальных генов (Прокофьева-Бельговская, 1986), подобно хромосомам 1, 9 и 16. Однако встречаются описания семейных случаев, когда при увеличенном блоке гетерохроматина наблюдали высокий рост и нарушения поведения (Dorus, 1978), а также сообщения о делениях Уq (Fitch et al., 1985). Известны также различные формы полисомии хромосомы У, при которых нередко наблюдают умственную отсталость (Ion et al., 1994).
Полисомии У. Полисомии У встречаются в виде дисомии У с кариотипом 47,ХУУ и трисомии У с кариотипом 48,ХУУУ. Эти два синдрома достаточно хорошо изучены еще в 70-х гг. Особенно следует отметить полисомию с кариотипом 48,ХХУУ—вариант синдрома Клайн-фельтера, как первую описанную форму полисомии У (Muldal, Ockey, 1960). Фенотипически эта форма не отличается от синдрома Клайнфельтера. Дисомия У. Клинически при синдроме дисомии У (кариотип 47,ХУУ) отсутствуют обязательные признаки, которые характерны для других гоносомных синдромов, но среди необязательных признаков непременно следует отметить высокий рост, нарушения поведения, физические аномалии, умственную отсталость, нарушение половой дифференцировки (Давиденкова и др., 1973; Kitsiou, Bartsocas,1986; Fryns et al., 1995). Частота встречаемости этого синдрома 1,5 на 1000 новорожденных. Цитогенетическими исследованиями показано, что у взрослых индивидуумов мужского пола с ростом выше 196 см и выраженными отклонениями в поведении каждый четвертый имел кариотип 47,ХУУ. При обследовании семейных случаев индивидуумы мужского пола с нормальным кариотипом, в том числе и дети, и с дисомией У имели резкие ростовые отличия в пользу последних. Нарушения поведения у многих больных проявляются в тех или иных агрессивных, иногда антиобщественных поступках. У больных с добавочной хромосомой У обнаруживают психопатические черты характера, импульсивность, отсутствие сильных привязаностей, плохое владение собой по поводу примитивных эмоций. Многие дети, страдающие этим заболеванием, растут и воспитываются в семьях, но они всегда склонны к неправильному поведению и антисоциальным поступкам. Показано также, что большинство опубликованных случаев синдрома дисомии У обнаруживают в психиатрических больницах или лечебно-профилактических учреждениях как детского, так и взрослого профиля для содержания особо опасных для общества лиц, а также в тюрьмах. У некоторых больных наблюдают шизофрению, депрессивные психозы, тяжелые формы психопатий и эпилепсии. Среди физических аномалий наблюдают макроцефалию, увеличение конечностей, прогнатизм, выступающие надбровные дуги, высокое небо, гипертрофию языка, грубые черты лица. Сочетание всех этих признаков создает впечатление больного, страдающего акромегалией. При данном синдроме наблюдают снижение интеллекта, у многих отмечают легкую олигофрению. В отдельных случаях умственная отсталость отягощается психопатизацией личности. У многих больных отмечают нарушение половой дифференцировки (крипторхизм, гипогонадизм, дисплазия гениталий). В большинстве своем мужчины, страдающие этим синдромом, фертильны. Дети, рождающиеся от этих мужчин, в редком случае имеют нормальный кариотип. Трисомии У. Встречаются единичные сообщения о полной форме трисомии У — кариотип 48.ХУУУ (Ridler et al.,1973). При этом обнаруживают умственную отсталость, поперечную складку на ладонях, гипоплазию зубной эмали, паховые грыжи, неопущение яичек, аномалии крупных сосудов (стеноз легочной артерии). Кроме того, описаны случаи мозаицизма 47,ХУУ/48,ХУУУ (Teyssier, Pousset, 1994), куда входит и сообщение о заключенном из австралийской тюрьмы, осужденном за убийство, а также случаи с кариотипом 49,ХУУУУ у детей до семи лет—глубоких инвалидов с грубыми неврологическими и психологическими расстройствами. В целом каждый подобный случай до настоящего времени нуждается в тщательном изучении и описании. ТРИПЛОИДИИ ХРОМОСОМ. Впервые живорожденный ребенок с полной формой триплои-дии хромосом (кариотип 69,ХХУ или 69,ХХХ) был описан в конце 60-х годов; до этого времени было известно только о мозаичных формах синдрома, когда наряду с триплоидным присутствовал и диплоидный клеточный клон. Популяционная частота синдрома неизвестна, несмотря на то, что описано около 100 случаев вместе с мозаичными формами. Беременность таким плодом осложняется многоводием и токсикозом второй половины, дети рождаются срезкой пренатальной гипоплазией (масса тела при рождении не превышает 1800 г) при беременности 34-35 недель. Продолжительность жизни детей до 6-7 месяцев (Faix et al., 1984). При данном синдроме наблюдают следующие признаки: микрофтальмия, колобома радужки, гипертелоризм, расщелина губы и неба, низко расположенные ушные раковины, микрогения, синдактилия кистей и стоп, искривление пальцев кистей и стоп, косолапость, гипоспадия или эписпа-дия, гипоплазия полового члена, аплазия наружных половых органов, крипторхизм. Из аномалий внутренних органов известны пороки сердца и крупных сосудов (дефекты перегородок и крупных сосудов), мочевой системы (кистозная дисплазия почек, гидроуре-тер, гидронефроз), желудочно-кишечного тракта (нарушения поворота кишечника, гипоплазия и агенезия желчного пузыря), желез внутренней секреции (гипоплазия надпочечников). Но самым основным проявлением этого синдрома являются пороки головного мозга, которые часты и специфичны. Самыми частыми из них являются спинномозговые грыжи (Лазюк, 1991), которые при других хромосомных синдромах практически не встречаются (исключение — синдром Эдвардса). При триплоидии также с высокой частотой встречаются пороки прозэнцефалической серии (Royston, Bannigan, 1987). Часто прозэнцефалия сочетается с гидроцефалией, что не встречается ни при одном хромосомном синдроме. Отмечают также цикло-пию и цебоцефалию, встречаются агенизия мозолистого тела, лиссэнцефалия, аплазия или гипоплазия мозжечка. Триплоидия хромосом считается единственной формой плоидности, совместимой с той или иной продолжительностью жизнью (Лазюк, 1991). Известно также не одно сообщение о живорожденном ребенке с тетрасомией (Pittetal., 1981; Teyssier etal.,1997). МАРКЕРНЫЕ ХРОМОСОМЫ И МОЗАИЧНЫЕ ФОРМЫ РАЗЛИЧНЫХ ХРОМОСОМНЫХ СИНДРОМОВ. Суммируя сообщения об известных хромосомных синдромах и аномалиях, следует сказать, что затруднений в цитогенетической диагностике они, в основном, не вызывают. Однако, как отмечено выше, известны случаи, когда методы классической цитогенетической диагностики мало эффективны, а подчас бессильны. При этом в первую очередь следует отметить добавочные (дополнительные) мини-хромосомы в кариотипе. Частота встречаемости маркерных хромосом определяется как 1-2 случая на 1000 при проведении пре- и пост-натальной цитогенетической диагностики (Buckton et al., 1985; Tozzi et al., 1988). Многочисленные исследования показывают, что маркерные хромосомы могут сегрегировать в семье, вызывая у одних членов умственную отсталость и ВПР, т.е. признаки хромосомной аномалии или синдрома, в то время как другие остаются фенотипи-чески нормальны и умственно сохранны. Как правило, дополнительную в кариотипе (маленьких размеров) маркерную хромосому невозможно идентифицировать традиционно-цитогенетическими методами, применяя даже высокоразрешающие способы определения. А между тем, анализ их происхождения и причастность к той или иной хромосомной аномалии или синдрому необходим для планирования деторождения в семье, для тактики проведения пренатальной диагностики (Ворсанова и др., 1990; 1991а; Зерова-Любимова, 1992; Vorsanova et al, 1994a,b). В данных случаях только применение методов молекулярно-цитогенетической диагностики позволяет определить происхождение маркерной хромосомы, ее хромосомоспе-цифичность и правильно ориентировать семью при генетическом консультировании (Buckton et al., 1985; McDermid et al., 1986). Мозаичные формы различных хромосомных синдромов также нуждаются в проведении молекулярной диагностики, особенно когда речь идет о небольшой доле аномальных клеток в организме больного (до 20%). Причем, довольно часто, лишь при использовании моле- кулярных методов удается обнаружить мозаичные формы заболевания, в то время как после стандартного цитогенетического исследования обнаруживается нормальный кариотип при клиническом проявлении синдрома. Такое уточнение диагноза необходимо для ведения больного, особенно в случаях гоносомных синдромов, в плане своевременной гормональной коррекции. Кроме того, диагностика мозаичных форм такого гоносомного синдрома, как синдром Тернера, при незначительной доле аномального клона (15-20%) клеток требует более тщательных молекулярных исследований для генетического консультирования в дальнейшем. Трудности при генетическом консультировании обусловлены тем, что такие больные чаще способны воспроизводить потомство по сравнению с пациентами, страдающими полной формой заболевания. А между тем, женщины с мозаицизмом по половым хромосомам имеют высокий риск рождения ребенка с ВПР и задержкой психомоторного развития (Бужиевская, Выговская, 1990). Кроме того, прослеживается наследственная предрасположенность к нерасхождению половых хромосом, что подтверждает также необходимость тщательного молекулярно-цитогенетического обследования детей с клиническими признаками гоносомного синдрома и их родителей (даже при нормальном кариотипе). Молекулярно-цитогенетическая диагностика помогает также разобраться в сложных случаях хромосомной патологии, такой как транслокации с участием нескольких хромосом, а также несбалансированные и сбалансированные транслокации с клинической картиной хромосомных синдромов, делеции и изохромосомы с атипичными фенотипическими проявлениями. В качестве примера мы приводим описания наиболее интересных случаев из практики лаборатории молекулярной цитогенетики нервно-психических заболеваний Московского НИИ педиатрии и детской хирургии Минздрава России, а также Центра клинической морфологии и генетики Ростовского государственного медицинского университета Минздрава России, где необходимость применения молекулярно-цитогенетической диагностики была очевидна.   Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право... 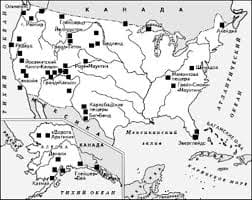 Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования...  Конфликты в семейной жизни. Как это изменить? Редкий брак и взаимоотношения существуют без конфликтов и напряженности. Через это проходят все... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|