
|
|
Клініка найбільш поширених мітохондропатій.Стр 1 из 5Следующая ⇒ Мітохондріальні хвороби Лекція № 4 Загальна характеристика мітохондріальної патології. Клініка, діагностика, лікування. (2 години) План 1. Характеристика мітохондріального геному. 2. Етіопатогенез мітохондріальних захворювань. 3. Класифікація мітохондропатій. 4. Клініка найбільш поширених мітохондропатій. 5. Принципи лікування мітохондропатій. Актуальність теми Серед сучасних спадкових хвороб, які зустрічаються у людей, існує безліч нозологічних одиниць, які повністю ще не ідентифіковані. Вони можуть ховатися за маскою будь-яких відомих хвороб, їх можуть лікувати як знайомі хвороби, але насправді вони мають багато відрізнень від описаних раніш хвороб, в тому числі і в відсутності ефекту від лікування. Такими хворобами до недавнього часу були і мітохондріальні хвороби. Після того, як був описаний мітохондріальний геном, встановлений тип успадкування і шукаються підходи до етіопатогенетичного лікування мітохондріальних хвороб, їх признали як окремий нозологічний клас. Крім того, після їх описання встановили, що вони не такі рідкі, як вважалося спочатку, і спостерігається певний ріст таких захворювань.
Навчальні цілі та мотивація Мітохондропатії займають високу питому вагу серед здорового населення 1,6%, а серед хворих із множинним одночасним ураженням нервової, скелетної, респіраторної, серцево-судинної систем порушення енергетичного обміну зустрічається у 45%. Мітохондропатії вкрай рідко діагностуються Зміст лекційного матеріалу Мітохондії — одні з найбільших органел клітини, які містяться в усіх евкаріотичних клітинах, окрім еритроцитів і зрілих кератиноцитів. Вони виконують функцію метаболічного центру та відповідають за виробництво енергії у вигляді АТФ. Накопичена енергія в подальшому трансформується в механічну (в м'язових клітинах), біоелектричну (у нервових клітинах). Найбільша кількість мітохондрій спостерігається переважно в клітинах, які споживають велику кількість енергії, наприклад у клітинах скелетних м'язів і серцевого м'яза, в екзокринних клітинах підшлункової залози, в овоцитах. У сперматозоїдах їх всього 4. Мітохондрії мають свої особисті ДНК, РНК і рибосоми, самі синтезують частину своїх білків, розмножуються шляхом поділу надвоє. Мітохондріям необхідні білки, які кодуються генами нуклеарних хромосом. ДНК мітохондрій являє собою дволанцюгову кільцеву молекулу розміром 16599 пар нуклеотидів. ДНК мітохондрії людини має 37 генів. Більшість генів ДНК мітохондрій кодують поліпептидні ланцюги, що беруть участь у ферментативному комплексі окисного фосфорилювання. Кожна клітина людського організму містить сотні мітохондрій і тисячі ДНК мітохондрій. Якщо мутація виникає в одній з молекул ДНК мітохондрій, то утворюється внутрішньоклітинна суміш мутантних і нормальних молекул. Таке явище називається гетероплазмією. Явище клітинного розподілу мутантних і нормальних мітохондрій називається реплікаційною сегрегацією.
Особливості мітохондріальної ДНК: 1. Строго материнський характер успадкування ДНК, тобто вони передаються від матері до дочок та синів. Сини мітохондріальної ДНК не передають. 2. Відсутня комбінативна мінливість - мітохондріальна ДНК належить тільки одному з батьків, отже, рекомбінаційні події відсутні, а нуклеотидна послідовність змінюється з покоління в покоління тільки за рахунок послідовного накопичення мутацій. 3. Мітохондріальна ДНК не має інтронів і ефективної ДНК репараційної системи. Це веде до збільшення частоти мутацій мітохондріальної ДНК порівняно з ядерною. 4. Усередині однієї клітини можуть одночасно співіснувати нормальні і мутантні мітохондріальні ДНК (явище гетероплазмії) Мітохондріальні захворювання - одна з найбільших класів дегенеративних хвороб нервово-м'язової системи, що має тяжкий проградієнтний перебіг, викликає виражену інвалідизацію і супроводжується резистентністю до лікування.
Класифікація мітохондропатій. Мітохондріальні хвороби класифікують за типом мутацій: 1.Місенс – мутантні мітохондріальні хвороби: - нейроофтальмопатія Лебера; - пігментний ретиніт. 2. Хвороби, які викликані мутаціями в генах транспортної РНК: - синдром MERRF; - синдром MELAS; 3. Мітохондріальні хвороби, які викликані делеціями або дуплікаціями мітохондріальної ДНК: - синдром Кернса-Сейра; - синдром Пірсона; - асиметричний птоз; - дилатаційна кардіоміопатія. 4. Хвороби, які викликані мутаціями, що знижують число копій мітохондріальної ДНК: - летальна інфантильна дихальна недостатність; - синдром молочного-кислого ацидозу. 5. Хвороби викликані мутаціями в ядерній ДНК: - фумарова ацидемія; - глутарова ацидемія.
Загальні клінічні риси МТХЗ: 1. Полісиндромність уражень з частим залученням нервової системи органа зору, серця і соматичних м'язів. 2. Більшість захворювань мають не вроджений характер і починаються в дитячому та молодому віці. 3. Проградієнтність перебігу з негативною динамікою і збільшенням симптомів ураження різних органів і систем. 4. Резистентність до традиційних методів лікування.
Сутність молекулярно-генетичних досліджень при мітохондропатіях. Мутації мітохондріальної ДНК визначаються у зразках м′язової тканини методом полімеразної ланцюгової реакції і гібридизації in situ. Виявлення виду мітохондріальної мутації з досить високим співвідношенням аномальної і нормальної мітохондріальної ДНК підтверджує діагноз мітохондріального захворювання. Відсутність мутації дає змогу припускати наявність патології, пов΄язаної з мутацією ядерної ДНК. Клініка найбільш поширених мітохондропатій. Синдром Лебера (спадкова атрофія зорових нервів) уперше описаний у 1971р. Теодором Лебером. Класична назва синдрому — «грім серед ясного неба». Захворювання проявляється у віці 6-62 роки. Першим проявом є зниження гостроти зору одного ока, через 7-8 тижнів – другого. Характерна неврологічна симптоматика: периферійна поліневропатія, тремор, атаксія, статичні парези, розумова відсталість. Також можуть бути наявні кістково-суглобні зміни: кіфоз, арахнодактилія. Перебіг захворювання прогресивний, однак можлива ремісія через 1-2 роки після початку захворювання. Диференціальну діагностику потрібно проводити із захворюваннями, що супроводжуються, зниженням гостроти зору: ретробульбарним невритом, краніофарингіомою, лейкодистрофіями. Критерії діагнозу: 1. Материнський тип успадкування; 2. Характерні клінічні ознаки; 3. Ідентифікація за допомогою молекулярно-генетичних досліджень мутацій в ДНК мітохондрій.
Мутації в генах тРНК Синдром MERRF (міоклонус-епілепсія, рвані червоні волокна", mioclonus-epilesy, rend red fibre). Уперше його описав N. Fukuhara із співавт. у 1980 p., хоча перші окремі публікації були зроблені P. Tsairis зі співавт., 1973; Y. Shapira, 1975. На успадкування захворювання по материнській лінії вказали Н.С. Rosing зі співавт., 1985; А.Е. Harding, I.J. Holt, 1988. Мутацію мтДНК виявили J.M. Sckoffner зі співавт., 1990. В Україні перша публікація належить О.Я. Гречаніній зі співавт. (1994). Синдром MERRF зумовлений точковими мутаціями в гені лізинової тРНК у позиціях 8344 і 8356 мтДНК. Захворювання успадковується із внутрішньосімейним поліморфізмом, у попередніх поколіннях можливі випадки захворювання з різним ступенем клінічних проявів, що може бути спричинено різним співвідношенням мутантних і нормальних мтДНК у різних овоцитах. Вік хворого на початку захворювання — від 3 до 65 років. Ранніми клінічними ознаками є швидка втомлюваність під час фізичних навантажень, біль у литкових м'язах, погіршення пам'яті, зниження уваги. Найтиповішим є симптомокомплекс: прогресивна міоклонус-епілепсія, що включає міоклонус (раптове, швидке, короткочасне м'язове скорочення, зумовлене втягуванням у патологічний процес ЦНС), атаксія і деменція. Також у хворих спостерігають генералізовані тоніко-клонічні судоми, нейросенсорну глухоту, атрофію зорових нервів, помірні ознаки міопатії, сенсорні порушення (розлад вібраційної чутливості і м'язово-суглобного відчуття) та інші неврологічні симптоми (відсутність сухожилкових рефлексів). Імовірний розвиток ліпоматозу. Перебіг захворювання прогресивний. Розвиток синдрому також варіабельний. Даних для встановлення прогнозу ще недостатньо, але відомо, що летальний кінець настає через 3—30 років від початку реєстрації перших клінічних проявів. Можлива трансформація синдрому MERRF у синдром MELAS, який також спричинений дефектом мітохондріального дихального ланцюга. Критерії діагнозу: — материнський тип успадкування; — початок захворювання у віці 3—65 років; — ЦНС — міоклонус, атаксія, деменція у поєднанні з нейросенсорною глухотою, атрофією зорових нервів, порушенням глибокої чутливості; — помірне підвищення рівня білка в лікворі; — недостатність 1, 3, 4-го комплексів дихального ланцюга; — ЕЕГ — генералізовані комплекси "спайк-хвиль"; — ЕМГ — первинно-м'язовий тип ураження; — КТ — атрофія мозку, лейкоенцефалопатія, кальцифікація базальних гангліїв (іноді); — "рвані червоні волокна" в біоптатах скелетних м'язів;
Диференціальну діагностику проводять з іншими прогресивними міокло- нус-епілепсіями (хворобою Ґоше, синдромом міоклонуса з нирковою недостатністю та ін.),хворобами накопичення (хвороба Краббе), з іншими дисгенезіями мозку.
Синдром MELAS (мітохондріальна енцефалопатія, лактат-ацидоз, інсуль-топодібні епізоди). Уперше його виділив у нозологічно самостійну форму S.G. Pavlakis зі співавт. у 1984 р. В основі патогенезу синдрому MELAS лежать точкові мутації мтДНК (у нуклеотидах 3243, 3271), причому існує кореляція між ступенем мутації і характером перебігу захворювання. Імовірно, що для прояву захворювання необхідне накопичення певної кількості мутантної мтДНК (56—95 %), однак в одній родині рідко народжуються 2 дитини з класичним варіантом хвороби. Перші ознаки захворювання з'являються, як правило, у 6—10 років (імовірні варіанти від 2 до 40 років). До прояву хвороби 90—100 % хворих розвиваються нормально. Частими початковими клінічними симптомами є судоми, рецидивний головний біль, блювання, анорексія. Одним із важливих симптомів мітохондріальної патології є непереносимість фізичних навантажень (різке погіршення самопочуття, поява м'язової слабкості, міалгії). Інсультоподібні епізоди проявляються рецидивними нападами головного болю, запамороченням, розвитком вогнищевої неврологічної симптоматики, коматозними станами. Причиною таких метаболічних інсультів є гостра недостатність енергетичних субстратів у клітинах, а також висока чутливість судин мозку до токсичних впливів. Провокаційними чинниками є лихоманка, інтеркурентні інфекції. Судоми — також один із головних маніфестних симптомів синдрому MELAS, однак вони дуже варіабельні: фокальні пароксизми, генералізовані тоніко-клонічні напади, міоклонії. Такі епілептичні напади резистентні до ан-тиконвульсантної терапії. З перебігом хвороби розвивається деменція. Можливе ураження ендокринної (цукровий діабет, гіпопаратиреоз) і серцево-судинної (передсердно-шлуночкова блокада) систем. Також відзначають низькорослість, порушення зору, атрофію зорових нервів, лихоманку, мозочковий синдром, синдром Вольфа—Паркінсона—Вайта, порушення серцевої провідності, прогресивну зовнішню офтальмоплегію, цукровий діабет. У хворих із синдромом MELAS відзначають додаткове ураження 1 і 4; 3 і 4; 1, 3 і 4; 1, 2, 3, 4 комплексів дихального ланцюга. Перебіг хвороби прогресивний. За раннього початку хвороби перебіг більш злоякісний.
Критерії діагнозу: — материнський тип успадкування; — вік маніфестації — до 40 років; — мігренеподібний головний біль із нудотою і блюванням; — інсультоподібні епізоди; — судоми; — у крові — лактат-ацидоз; — у сечі — органічна ацидурія; — КТ — кальцифікація базальних гангліїв; — наявність "рваних червоних волокон" у біоптатах скелетних м'язів; — прогресивний перебіг. Синдром MELAS слід диференціювати з іншими мітохондріальними хворобами, синдромом Лея (підгостра некротизивна енцефалопатія), органічними ацидеміями, гомоцистинурією, синдромом Фабрі, вродженими вадами серця, судинними аномаліями.   ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между... 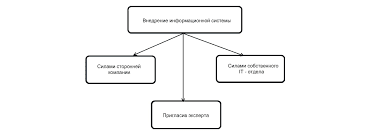 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)... 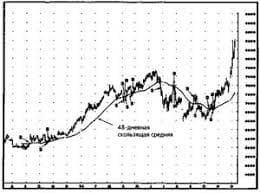 Что вызывает тренды на фондовых и товарных рынках Объяснение теории грузового поезда Первые 17 лет моих рыночных исследований сводились к попыткам вычислить, когда этот...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|