
|
|
Ідіопатична тромбоцитопенічна пурпураВизначення. Ідіопатична тромбоцитопенічна пурпура (ІТП), (симптомокомплекс Верльгофа) – первинна тромбоцитопенія з аутоімунним механізмом утворення антитромбоцитарних антитіл. Патогенез. Деструкція тромбоцитів, покритих аутоантитілами (імуноглобулінами класу G), виникає внаслідок їх фагоцитозу макрофагами в селезінці, в печінці, в обох органах. Середня тривалість життя тромбоцитів при ІТП нерідко скорочується до 15 годин, тоді як в нормі вона складає 220 годин. Антитіла можуть бути направлені як проти антигенів тромбоцитів, так і проти антигенів мегакаріоцитів. В останньому випадку кількість мегакаріоцитів в кістковому мозку хворих понижується. Класифікація (А.Ф. Романова, 1997). За перебігом розрізняють 2 форми ІТП: 1. Гостра – з раптовим початком в дітей від 2 до 6 років та виздоровленням в терміни до 6 місяців. 2. Хронічна – із прихованим початком, рідкими спонтанними ремісіями і частими рецидивами. Пік захворюваності у віці від 20 до 40 років. Приклад формулювання діагнозу: Ідіопатична тромбоцитопенічна пурпура, хронічне протікання, фаза рецидиву. Хронічна післягеморагічна залізодефіцитна анемія легкого ступеня важкості, гіпохромна, норморегенераторна, нормобластна. Клінічна картина. Хронічною формою ІТП хворіють переважно жінки (3:1). Кровоточивість носить мікроциркуляторний характер: крововиливи на шкірі у вигляді петехій і екхімозів поєднуються з носовими кровотечами, менорагіями. Гематурія, мелена, кривава рвота – нехарактерні. У 10-20% хворих відмічається помірна спленомегалія (частіше за даними УЗД). В периферичній крові після рясної кровотечі – картина гострої постгеморагічної анемії, при повторних епізодах – хронічна післягеморагічна анемія різного ступеня вираженості. В кістковому мозку – гіперплазія мегакаріоцитарного і еритроцитарного ростків. Діагностичні критерії. 1. Відсутність зв’язку кровоточивості з будь-яким попереднім чи фоновим захворюванням. 2. Геморагічний синдром з петехіально-екхімозним типом кровоточивості. 3. Тромбоцитопенія в периферичній крові (нижче 150-100*109/л – до одиничних тромбоцитів в препараті). 4. Позитивні “судинні” проби (джгута, щипка, манжеткова та ін.). 5. “Відпечаткові” крововиливи на шкірі (при носінні тісного одягу, поясів, після глибокої пальпації та ін.) 6. Подовження часу капілярної кровотечі (проба Д’юка) при нормальних показниках часу зсідання крові за Лі-Уайтом. 7. Порушення ретракції кров’яного згустка (зниження індексу менше 40%) 8. Виявлення антитромбоцитарних антитіл (за методом Діксена, за даними ІФА). 9. Підвищення рівня циркулюючих імунних комплексів (ЦІК), імуноглобулінів класу G. 10. Поєднання тромбоцитопенії з аутоімунною гемолітичною анемією (позитивна пряма проба Кумбса) – синдром Еванса-Фішера. Гетероїмунні форми тромбоцитопеній (тромбоцитопатій). Тромбоцитопенії, викликані вірусною інфекцією. Механізм руйнування тромбоцитів імунний: зміни антигенної структури тромбоцитів виникають під впливом вірусу. Кровоточивість (другий тип) появляється через 2-3 тижні після початку вірусної інфекції – краснухи, кору, вітряної віспи (у дітей), грипу, аденовірусної інфекції – в дорослих. Протікає гостро, крім шкірних проявів спостерігаються шлункові та ниркові кровотечі. У 62% пацієнтів – лімфаденопатія, збільшення селезінки (8%). Прогноз сприятливий. Зміни зникають через 2,5-4 тижні. Ефективним є лікування глюкокортикоїдами. Гемофілії. Визначення. Термін «гемофілія» може використовуватися як для визначення будь-якого дефіциту плазмових факторів зсідання крові, так і більш вузько - для найменування захворювань, обумовлених спадковими порушеннями коагуляційного гемостазу, перебігаючих з пониженням зсідання крові. Серед коагулопатій спадкового генезу найчастіше зустрічається дефіцит ф. VIII (гемофілія А) і ф. ІХ (гемофілія В): відповідно 68-80% і 8-15%. В деяких популяціях (євреї) дефіцит ф. ХІ (гемофілія С) реєструється частіше, ніж гемофілія А і В. Рідше спостерігається спадковий дефіцит ф. VII (гіпопроконвертинемія), який може не проявлятися епізодами кровоточивості в анамнезі, але маніфестувати тяжкими кровотечами в післяопераційному періоді. Етіологія і патогенез. Гемофілії А і В, обумовлені спадковим дефіцитом або молекулярними аномаліями VIII i IX факторів зсідання, передаються за рецесивним типом. Ген гемофілії локалізований в Х-хромосомі, в зв’язку з чим жінки є кондукторами цього захворювання, але не страждають кровоточивістю, так як при наявності другої нормальної Х-хромосоми активність факторів (VIII i IX) в них знижена в середньому вдвічі, в порівнянні з нормальними величинами. Крайнє рідко зустрічається аутосомно-домінантні форми гемофілії А, в тому числі і у жінок – “жіноча гемофілія”. При всіх поширених формах спадкових дефіцитів факторів зсідання крові (гемофілії А,В,С, хвороба Віллебранда) має місце ізольоване порушення першої фази внутрішнього механізму зсідання крові. При дефіциті ф. VII – порушений зовнішній механізм формування протромбіназної активності. В основі прогресування гемофілічних артропатій лежить розвиток синовіїтів, пов’язаних з геморагічним просяканням синовії, поверхневих хрящів з наступним гемосидерозом. Згідно із сучасними уявленнями гемосидероз є важливим фактором деструктивно-склеротичних змін в синовіальній оболонці, хрящах і суглобових поверхнях кісток. Класифікація. Класифікація спадкових порушень коагуляційного гемостазу, протікаючих з пониженням зсідання крові представлена в табл. 76. Рубрикація гемофілій за рівнем дефіциту фактора до середньої норми поділяє їх на наступні форми: дуже важка - 0-1%; важка – 1-2%; середньої важкості - 2-5%; легка - більше 5%. Спонтанні гемофілії спостерігаються при рівні антигемофільного фактора нижче 5%, тоді, як при концентрації вище 5% реєструються лиш посттравматичні і післяопераційні кровотечі, які можуть бути досить рясними, не тільки при важких, але і при “легких” формах хвороби і при відсутності замісної терапії призводить до летального наслідку.
Приклад формулювання діагнозу: Гемофілія А, важка форма. Гострий гемартроз лівого колінного суглобу. Гемофілічна артропатія правого колінного суглобу. Гостра післягеморагічна анемія середнього ступеня важкості. Клінічна картина. Клінічні прояви гемофілій А і В однакові. Тяжкі форми діагностуються при народженні: значні кефалогематоми, підшкірні крововиливи, пізні кровотечі з пупкового канатика. При легких формах та формах середньої важкості захворювання розпізнається на 2-3 році життя: випадкові порізи, травми, прикуси язика супроводжуються рясними кровотечами. В подальшому в клініці домінують прояви гемофілічних артропатій. Гострі гемартрози проявляються: при важких формах з 2-3-річного віку, при середній важкості – з 4-6 років. Найчастіше уражаються колінні, ліктьові і гомілково-ступневі, рідше – плечові, променево-зап’ясткові, кульшові суглоби. З віком важкість і поширеність суглобових змін неухильно прогресує і до 15-20 років у хворих на гемофілію розвивається складна дисфункція опорно-рухового апарату, що веде до інвалідизації. Діагностичні критерії. 1. Сімейний анамнез (в 1/3 хворих вказівки на геморагічні захворювання у родичів можуть бути відсутні – гемофілія розвивається в результаті мутації гена) 2. Гематомний (І) тип кровоточивості. 3. Гемофілічні артропатії: поєднання гострих гемартрозів (первинних та рецидивуючих) з хронічними деструктивними остеоартрозами. 4. Раннє визначення м’якотканнинних змін (гіперплазії і потовщення), змін периартрикулярних тканин, накопичення крові в суглобовій сумці і її кишенях, гемосидероз синовіальної оболонки, тощо методами променевої діагностики (УЗІ, МР – томографії). 5. Гематурія в поєднанні з приступами ниркової коліки (утворення згустків в сечових шляхах - у 15 – 30% хворих. 6. Продовження часу зсідання крові по Лі – Уайту при нормальних показниках часу кровотечі за Д’юком, кількості тромбоцитів, протромбінового часу (при гемофіліях А і В). 7. Встановлення дефіциту фактора зсідання крові і його кількісне визначення по нормалізації часу зсідання після змішування плазми хворого з одногрупною плазмою здорової людини в корекційних пробах. 8. Вторинний ревматоїдний синдром – симетричне ураження як крупних, так і дрібних суглобів (в тому числі і тих, які не вражалися крововиливами): - ранкова скутість; - постійність больового синдрому; - загострення після замісної терапії; - підвищення в плазмі рівня ревматоїдного фактору, СРБ, сіалових кислот, гама-глобулінів та ін.; - рентгенологічні ознаки ревматоїдного артриту (звуження суглобових щілин, узурація суглобових поверхонь та ін.)   Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам...  ЧТО ПРОИСХОДИТ, КОГДА МЫ ССОРИМСЯ Не понимая различий, существующих между мужчинами и женщинами, очень легко довести дело до ссоры... 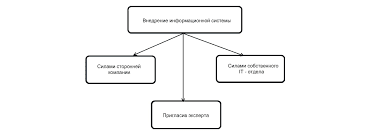 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|