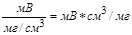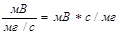|
|
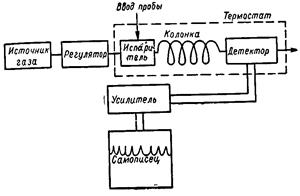
ГЛАВА 4. ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗСпособность атомов и молекул поглощать энергию, поступающую к ним извне, вызывает их возбуждение. Избыточная энергия атомов или молекул, полученная при возбуждении, может быть израсходована на ионизацию вещества, на фотохимические реакции, на нагрев вещества. Кроме того, возбужденные атомы или молекулы способны отдавать избыточную энергию в виде света. Большинство твердых веществ при сильном нагревании светятся, т. е. наблюдается тепловое излучение раскаленного тела. Естественно, что чем больше энергии при данной температуре поглощает тело, тем больше оно ее излучает. У некоторых веществ наблюдается холодное свечение — люминесценция т. е. свечение при комнатной температуре без нагревания. Это излучение является неравновесным и продолжается долгое время после прекращения действия внешнего возбуждающего фактора. Люминесценцией называется избыток теплоты над температурным излучением в случае, если это избыточное излучение обладает конечной длительностью примерно от 10-10 с и более. В зависимости от вида люминесценции обычно рассматриваются следующие разделы люминесцентного анализа: 1) флуоресценция, основанная на свечении вещества при поглощении световой энергии; 2) катодолюминесценция, вызванная бомбардировкой быстрых электронов; 3) хемилюминесценция — свечение, вызванное протеканием специфических химических процессов. Все люминесцирующие вещества объединяются под общим названием люминофоры. Органические и неорганические люминофоры существенно различаются по природе свечения. Известны два механизма возникновения свечения: 1) свечение отдельных центров, когда процесс возникновения люминесценции протекает лишь в одной частице, являющейся как поглотителем энергии, так и излучателем световых квантов; 2) рекомбинационные процессы свечения, при которых поглощение энергии осуществляется не теми частицами, которые излучают световые кванты. По первому механизму осуществляется свечение большинства органических веществ в растворах, а также внутрикомплексных соединений органических люминесцентных реагентов с катионами. По второму механизму осуществляется свечение нафталина, антрацена и их производных, а также сульфида цинка, сульфида кадмия, окиси кальция и т. п. Во всех видах люминесценции проявляются характерные свойства веществ, что может служить основой для их распознавания и изучения, т. е. что составляет предмет санитарно-химического анализа. В практике наиболее широкое применение получила и, по-видимому, получит дальнейшее развитие флуоресценция — свечение анализируемых растворов в УФ-свете. Сортовой анализ широко применяется для различения многих органических препаратов, для выявления суррогатов, при сортировке различного вида топлив и т. д. Люминесцентный химический анализ обладает исключительной чувствительностью, что является незаменимым качеством для целей санитарно-химического анализа: люминесценцию можно наблюдать при очень малых концентрациях люминесцирующего вещества. Методика выполнения люминесцентных реакций — микрохимическая (капельная). Наличие искомого вещества устанавливают либо по появлению люминесценции, либо по ее тушению, полному или частичному. Люминесцентные реакции во многих случаях не требуют разделения смеси и выделения искомого вещества. В качественном люминесцентном анализе для определения неорганических и органических веществ используют собственную люминесценцию. Из неорганических веществ в растворенном состоянии в УФ-свете люминесцируют соли лактаноидов, соли уранила, соли тяжелых металлов Tl+, Sn'2+, Sb?+, Bi§+, Pb2+, In3* и др. Из органических веществ собственной люминесцен-цией обладают битумы, смолы, смазочные масла, многие витамины, порфирин адреналин и целый ряд канцерогенов. Анализ может быть основан на изменении люминесценции реактива под влиянием искомого вещества. Образование соединений многих, не люминесцирующих в водных растворах, катионов с молекулой органического реагента сопровождается изменением или появлением люминесценции этого реагента. Ион натрия с цинкуран ил ацетатом дает зелено-желтую люминесценцию. Бериллий с морином образует комплекс, люминесцирующий ярко-зеленым цветом. Многие катионы с оксихинолином дают соединения, обладающие характерной люминесценцией. Кристаллофосфоры (вещества, в возникновении флуоресценции которых принимает участие весь кристалл) на основе окиси кальция образуют многие элементы с характерной люминесценцией. Задача качественного анализа усложняется, когда смесь состоит из нескольких люминесцирующих веществ; в этом случае применяют светофильтры или сочетание люминесцентного анализа с хроматографическим. Наиболее избирательные методы анализа основаны на спектральном разложении света люминесценции и изучения спектральных характеристик люминесценции спектрофотометрическим методом. Количественный люминесцентный анализ — флуометрия — основан на зависимости между интенсивностью люминесценции и концентрацией анализируемого вещества, т. е. на пропорциональности интенсивности люминесценции количеству поглощающих и излучающих центров и доле поглощенного света. Чувствителыюсть флуометрических методов значительно выше, чем фотометрических, что очень важно для целей санитарно-химического анализа. Главным условием успешного применения люминесцентных реакций для количественного анализа является наиболее полное превращение поглощенной энергии в люминесцентное излучение. Флуометрические измерения выполняются как визуально, так и с помощью аппаратурных методов регистрации возникающего излучения. Глава 5. ГАЗОВАЯ ХРОМАТОГРАФИЯ Предмет газовой хроматографии. Газовая хроматография (ГХ) представляет собой метод разделения летучих соединений, основанный на распределении вещества между двумя фазами, одна из которых неподвижная (стационарная) с большой поверхностью, а другая — газ, протекающий через неподвижную фазу. В случае газоадсорбционной хроматографии стационарная фаза твердая. При этом адсорбционными наполнителями колонок служат силикагель, молекулярные сита, активированный уголь. В случае газожидкостной хроматографии стационарная фаза — жидкость (ГЖХ). В этом случае основой разделения является процесс распределения вещества пробы между тонкой пленкой жидкости, нанесенной на поверхность инертного твердого носителя, и газовой фазой. Достоинства газовой хроматографии — высокая чувствительность, относительная скорость, точность и простота. Аппаратурное оформление. Газовый хроматограф (рис. 59) состоит из баллона с газом-носителем 1 (азот, гелий), регулятора расхода газа-носителя 2, испарителя — системы ввода пробы 3, термостатов 4, термостатируемой колонки 5, детектора 6 и самописца 7.
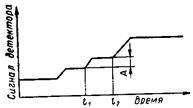
Рис. 59. Схема хроматографа. Сущность метода. В газожидкостной хроматографии разделяемые компоненты перемещаются по колонке с помощью газа-носителя, распределяясь между ним и стационарной фазой, нанесенной на твердый носитель определенной степени зернения. Компоненты смеси в соответствии со своими коэффициентами распределения селективно удерживаются стационарной фазой до тех пор, пока не образуют отдельных зон в газе-носителе. Эти зоны выносятся из колонки газом-носителем и регистрируются детектором в виде сигналов, являющихся функцией времени. Преимущества метода ГЖХ: колонка постоянно регенерируется газом-носителем; компоненты смесей разделяются полностью, не смешиваясь друг с другом; время анализа невелико. Недостаток метода ГЖХ: компоненты сильно адсорбируются колонкой, выходят медленно либо не выходят вообще. Этот недостаток можно преодолеть, используя программирование температуры колонки, т. е. повышение температуры последней на протяжении всего процесса анализа. Запись результатов хроматографического анализа на диаграммной ленте называется хроматограммой (рис. 60). Ось ординат фиксирует сигнал детектора в милливольтах, а ось абсцисс— время.
Рис. 60. Хроматограмма. Характеристика метода. Чаще всего продолжительность ГЖХ измеряется минутами, т. е. очень мала вследствие быстрого установления равновесия между стационарной и подвижной фазами, что позволяет использовать высокие скорости газа-носителя. Газовая хроматография дает возможность разделять соединения с практически одинаковыми температурами кипения за счет использования селективных стационарных фаз. Для газохроматографического процесса характерный параметр — время удерживания, т. е. время от момента ввода пробы до выхода максимума пика. При контроле температуры термостата и скорости потока воспроизводимость времени удерживания около 1%, а поэтому оно может использоваться для идентификации каждого пика. Несколько соединений могут иметь одинаковые или близкие значения времени удерживания, однако каждое соединение имеет только одно определенное время удерживания. В ряде случаев оно не зависит от присутствия других компонентов, например, при анализе водных растворов ряд авторов наблюдали значительное изменение времени удерживания компонентов, элюируемых после высокополярного соединения, присутствующего в смеси в большой концентрации. Область применения метода. Для санитарно-химического анализа важна количественная характеристика метода ГЖХ: площадь каждого пика хроматограммы пропорциональна концентрации соответствующего компонента, следовательно, она может быть использована для точного определения этой концентрации. Точность, достигаемая в ГЖХ, зависит от методики проведения опыта, конструкции детектора, метода интегрирования и концентрации пробы. Относительная ошибка в определении площадей основных пиков хроматограммы, измеренных вручную, обычно составляет около 1—2%. Метод ГХ высокочувствителен. Простейшие конструкции детектора по теплопроводности позволяют определять около 0,01% компонента в смеси. Пламенно-ионизационный детектор дает возможность легко определять миллионные доли компонента, а селективный детектор по электронному захвату и фосфорный детектор — миллиардные доли и даже пикограммы (10-12 г) вещества, причем для ГХ требуются небольшие пробы. Этот метод применяется для идентификации и определения любого вещества, имеющего давление пара 1—1000 мм при температуре колонки (—70)—(+400)° С.
Хроматографический процесс. Газ-носитель поступает в термостатируемую хроматографическую колонку из баллона. При изотермическом режиме анализа сопротивление колонки постоянно. Для поддержания постоянного давления на входе в колонку и для сохранения постоянной скорости газа-носителя используют регулятор давления. При данной температуре постоянная скорость газа-носителя обеспечивает постоянство времени удерживания анализируемых веществ. В связи с постоянством скорости газа-носителя компоненты анализируемой смеси можно охарактеризовать удерживаемым объемом (объемом газа-носителя, прошедшего за время от начала анализа до момента выхода пика). Требования, предъявляемые к газу-носителю: инертность к анализируемой пробе и жидкой фазе; обеспечение соответствующих диффузионных характеристик анализируемых веществ; легкодоступность и аналитическая частота; низкая стоимость; соответствие используемому детектору. Эффективность колонки зависит от выбора соответствующей линейной скорости. Для колонок с наружным диаметром 6 и 3 мм оптимальная скорость соответственно равна 75 и 25 мл/мин. Более просто и быстро скорость газа-носителя можно измерить пенным измерителем, который присоединяют к выходу хроматографа. В пенный измеритель скорости наливают раствор моющего вещества, заполняя резиновую грушу и трубку измерителя до уровня бокового отвода (но не выше). Затем добавляют еще немного раствора так, чтобы он попадал в боковой отвод только тогда, когда нажата резиновая груша. При протекании газа-носителя через измеритель скорости во время нажатия резиновой груши образуются мыльные пузыри, которые выносятся в градуированную трубку. Прежде чем приступить к измерениям, смачивают трубку, пропуская несколько таких пузырей. Определяя скорость потока, по секундомеру измеряют время t (в секундах), необходимое мыльному пузырю для прохождения объема 10 мл, заключенного между двумя отметками на градуированной трубке. Скорость потока газа-носителя рассчитывают по формуле
Ввод пробы - одна из самых ответственных процедур ГХ анализа является, особенно актуальной в ГХ санитарном анализе, где содержание определяемого ингредиента может быть очень низким. Проба должна быть введена на колонку быстро в виде «пробки», т. е. чтобы поступаемая проба была возможно менее разбавленной газом-носителем. Газы обычно вводят с помощью герметичных газовых шприцев, пробоотборных кранов или клапанных устройств. Дозирующее устройство должно обеспечивать, кроме точности и воспроизводимости дозируемого объема, еще и возможно меньшее время ввода пробы. Жидкие пробы вводят шприцами. В последнее время стали выпускать устройства для непосредственного ввода проб твердых веществ. Однако более простой метод ввода твердых проб состоит в растворении веществ в таком растворителе, пик которого легко отделяется от пиков веществ. Стандартная методика ввода проб состоит в прокалывании шприцем и введении определяемого объема пробы. Для ввода жидких проб применяются также дозирующие пипетки, в которых анализируемая жидкость заполняет под действием капиллярных сил дозирующий объем, после чего жидкая проба переносится в колонку. При анализе летучих веществ и для обеспечения высокой точности анализа прибегают к вводу в испаритель хроматографа стеклянных или металлических ампул, содержащих взвешенное количество вещества. Посредством специального устройства ампулу разрушают, и вещества поступают в колонку. Хроматографическая колонка — главная составная часть, в которой достигается действительное разделение компонентов смеси. Колонка может быть изготовлена из прямой, согнутой или свернутой в спираль медной, алюминиевой, стеклянной или из нержавеющей стали трубки. Следует ограничить изготовление колонок из меди, так как этот металл сильно адсорбирует или реагирует с аммиаком, ацетиленами и др. Успех ГХ зависит от выбора колонки. Для обеспечения равномерной набивки трубки сначала наполняют твердым инертным носителем, на который в виде тонкой пленки нанесена нелетучая жидкость, а затем скручивают в спираль для увеличения длины колонок. Капиллярные колонки — это полые трубки малою диаметра, на стенки которых нанесена тонкая пленка жидкости. Наиболее эффективными являются прямые колонки, однако при работе в области высоких температур они вызывают некоторые затруднения. При скручивании трубки в спираль диаметр спирали должен быть в десять раз больше диаметра трубки. Это условие обязательно для уменьшения влияния диффузии и стеночного эффекта. С увеличением длины колонки разделение улучшается. Однако, когда колонки слишком длинные, разделение становится хуже, при этом необходимо создавать очень высокие давления на входе, которые вызывают затруднения при вводе пробы и обеспечении герметичности. Длина обычных аналитических колонок составляет 1—4 м. Для длинных колонок характерно, что допустима величина пробы пропорциональна корню квадратному из длины колонки. Так как количество фазы в колонке пропорционально ее длине, то допустимая доза пропорциональна корню квадратному из количества фазы. В этом случае становится возможным увеличение проб, что является преимуществом длинных колонок.
Приготовление колонок. При нанесении жидкой фазы на твердый носитель могут быть использованы несколько методов. Мы разберем два наиболее распространенных метода, рекомендуемых для применения в практике санитарно-химических лабораторий. Метод роторного испарителя. Требуемое количество жидкой фазы, растворенное в соответствующем растворителе, помещают в круглодонную колбу роторного испарителя. Затем добавляют взвешенное количество твердого носителя. Приведя роторный испаритель в действие, удаляют растворитель в вакууме. Метод нанесения фазы в чашке. Взвешенное количество жидкой фазы, растворенной в определенном количестве растворителя, добавляют к взвешенному количеству твердого носителя, помещенному в фарфоровую чашку. Количество растворителя должно быть таким, чтобы только смочить твердый носитель. Растворитель испаряется самопроизвольно либо в воздушном потоке фена. Смесь во время сушки рекомендуется осторожно перемешивать медленным встряхиванием чашки. Перемешивать смесь другим способом не рекомендуется, так как это может привести к разрушению частиц твердого носителя.
Кондиционирование колонок. Условия кондиционирования: колонку необходимо прогреть в течение 2 ч и более при температуре на 25°С выше той, при которой колонка будет работать; однако температура прогрева должна быть ниже верхнего температурного предела для данной жидкой фазы. Во время кондиционирования колбу следует продувать слабым потоком газа-носителя (5—10 мл/мин). При этом рекомендуется отсоединить детектор во избежание его загрязнения. Объекты санитарно-гигиенического анализа — это сложные многокомпонентные смеси, которые часто трудно анализировать методом газовой хроматографии на обычных колонках. Поэтому одним из эффективных методов исследования состава этих смесей является капиллярная газовая хроматография. Однако успешное применение этого метода возможно только при соблюдении определенных требований к материалу колонки и способу модификации внутренней ее поверхности. Из-за сложности изготовления стальных капилляров и неудобства работы со стеклянными часто используют медные и латунные капиллярные колонки. Эти колонки при разделении полярных веществ характеризуются значительной каталитической, химической и адсорбционной активностью внутренней поверхности и, как следствие, трудностью изготовления из них высокоэффективных колонок. Для уменьшения этих явлений внутреннюю поверхность колонки обрабатывают водными растворами солей благородных металлов и бихромата калия, эпоксидной смолой; наносят на внутреннюю поверхность капилляра пористый слой инертного сорбента. Пригодность капиллярной колонки для нанесения неподвижной фазы определяют по ее адсорбционной активности. В качестве критерия активности используют сравнение коэффициентов ассиметрии газохроматографических пиков. Идентификацию компонентов выполняют с использованием индивидуальных углеводородов. Для определения состава неразделенных пиков хроматографируют исходную сложную смесь углеводородов и парафино-нафтеновую часть после сульфирования ароматических и олефиновых углеводородов.
Твердый носитель. Назначением твердого носителя является обеспечение большой, однородной и инертной поверхности для распределения жидкой фазы. Оптимальный твердый носитель должен обладать: инертностью, исключая тем самым адсорбцию и химическое взаимодействие; большой механической прочностью во избежание измельчения при заполнении колонки; большой удельной поверхностью (от 1 до 20м2/г); однородной формой и равномерными размерами частиц для обеспечения эффективной упаковки; воспроизводимостью (получением повторных партий носителя, не отличающихся заметно по своим основным свойствам). Пока неизвестен материал, удовлетворяющий всем перечисленным требованиям, однако в продаже имеются несколько подходящих твердых носителей. Сырьем для большинства твердых носителей, применяемых в ГХ, служит кизельгур. Ряд марок носителей промываетася кислотой, что позволяет удалить из поверхностного слоя окиси металлов и тем самым снизить каталитическую активность. Кроме того, промывка кислотой удаляет из носителя пыль. Один из носителей - хроматон N получают кальцинированием кремнезема, очищенного физическим и химическим методами, с последующим формованием в шарики. Использованное сырье, внешний вид и свойства аналогичны хорошо известному хромосорбу W, а также целиту-545, выпускаемому фирмой «Johns Manville». Примерный химический состав: SiO2 (93%), А12О3 (3,3%), Fe2O3 (0,04%), ТiO2 (0,01%), CaO+MgO (0,1%), Na2О+K2О (3,4%).
Стационарные фазы. В ГЖХ наиболее важной задачей является правильный выбор неподвижной (стационарной) фазы. Идеальная жидкая фаза должна обладать следующими свойствами. 1. Коэффициенты распределения в ней анализируемых веществ различны. 2. Растворимость в ней анализируемых веществ находится в разумных пределах. 3. Упругость пара жидкой фазы при температуре анализа ничтожна. Именно возможность широкого выбора жидких фаз и определяет универсальность и селективность метода ГЖХ. Коэффициент распределения может изменяться в 50 раз для различных фаз. Это приводит к пятидесятикратным различиям в величинах времени удерживания, что способствует лучшему разделению. Количество жидкой фазы должно быть достаточным для того, чтобы покрыть частицы носителя тонким равномерным слоем. При избытке жидкой фазы происходит накапливание ее в промежутках между частицами и в результате снижается эффективность колонки Для носителей на основе диатомовых земель эффективность резко падает при нанесении жидкой фазы в количествах, превышающих 30% массы носителя. В настоящее время наблюдается тенденция к применению колонок с небольшим процентом жидкой фазы (2—10%),что сокращает время анализа. На тефлоновых носителях максимальное количество жидкой фазы составляет около 10%. Однако слишком малые количества неподвижной фазы могут прийти к тому, что активные центры поверхности носителя окажутся не покрытыми пленкой жидкости. Последнее может вызывать необратимую адсорбцию или разложение пробы. При выборе количества жидкой фазы необходимо иметь в виду летучесть пробы. Вещества с низкой летучестью лучше разделяются на колонках с небольшими количествами жидкой фазы 3% и менее. Для соединений с высокой летучестью (легкие углеводороды) требуются большие количества жидкой фазы (20—30%), так как растворимость их в жидких фазах мала. Чем больше количество жидкой фазы, тем более длительное время соединение находится в жидкости и тем лучше условия их разделения. Детекторы. Детектор регистрирует присутствие каждого компонента и позволяет измерять его количество в потоке, выходящем их хроматографической колонки. Детекторы могут быть интегральные и дифференциальные. Сигнал интегрального детектора пропорционален общей массе вещества в элюированной полосе. Когда через детектор проходит чистый газ-носитель, на диаграммной ленте записывается горизонтальная прямая линия. При прохождении полосы компонента перо самописца перемещается поперек диаграммной ленты на расстояние, пропорциональное массе компонента в полосе. При элюировании полосы следующего компонента перо снова перемещается перпендикулярно диаграммной ленте. Полученная хроматограмма (рис. 61) состоит из серии ступеней, высота которых пропорциональна массе компонента, соответствующего данной ступени. Примером интегрального детектора может служить бюретка для титрования. Сигнал дифференциального детектора пропорционален концентрации или массовой скорости потока элюируемого компонента. Примером концентрационного детектора является детектор по теплопроводности — катарометр, а примером потокового детектора — пламенно-ионизационный детектор. Хроматограмма, полученная при использовании дифференциального детектора, состоит из серии пиков, соответствующих отдельным компонентам анализируемой смеси веществ. Площадь каждого пика пропорциональна массе соответствующего компонента -это легко доказывается математически (рис. 63).
Рис. 61. Интегральная хроматограмма. А — расстояние, пропорциональное массе компонента, элюированного за интервал времени t2—t1.
Рис. 62. Дифференциальная хроматограмма. S — площадь, пропорциональная массе компонента, элюированного за интервал времени t2—t1 Дифференциальные детекторы шире распространены, чем интегральные, вследствие их большей точности и удобства использования. Математическая интерпретация процессов, происходящих в потоковых детекторах, показывает, что в отличие от концентрационного детектора площадь пика для потокового детектора не зависит от скорости газа-носителя. Поэтому для пламенно-ионизационного детектора поддержание постоянной скорости не имеет такого значения, как в случае детектора по теплопроводности. Характеристики детектора. Существуют некоторые общие характеристики, позволяющие сравнивать различные детекторы. Это следующие характеристики: 1) чувствительность, 2) уровень шумов, 3) линейный диапазон, 4) природа сигнала Детектор должен быть нечувствительным к температурным изменениям и изменениям скорости газового потока. Универсальный детектор должен реагировать на соединения всех классов. Для санитарно-химического анализа могут потребоваться специальные детекторы (например, детектор по электронному захвату или фосфорный), обладающие селективной чувствительностью только к определенным классам соединений. Если принять для концентрационных детекторов за единицу чувствительности сигнал, равный 1 мВ при концентрации 1 мг на 1 см3 газа-носителя, то чувствительность детектора выражается в единицах:
Чувствительность детекторов по теплопроводности составляет 1000 — 10 000 мВ -см3/мг. Те же авторы для потоковых детекторов вывели следующую единицу чувствительности:
Необходимо отметить, что чувствительность для концентрационного детектора является функцией параметров газохроматографического процесса и пропорциональна скорости газового потока: А = (SC1C2C3) w, где А — чувствительность детектора, мВ -см3/кг; S — площадь пика, см2; С1 — чувствительность самописца, мВ/см диаграммной ленты, С2 — обратная величина скорости диаграммной ленты, мин/см; С3 — скорость газа-носителя, мл/мин; w — масса компонента, мг. Для потоковых детекторов чувствительность не зависит от скорости газового потока и изображается функцией параметров газохроматографического процесса - а1 = (SС1С2) w. Выходной сигнал детектора можно увеличить до любой желаемой величины с помощью электронного усиления, т. е. можно получить сколь угодно большею чувствительность детектора. Однако из-за собственных шумов детектора и электронного прибора может наступить момент, когда уровень шумов последних перекроет сигнал детектора. Вследствие этого уровень шумов ограничивает минимальную концентрацию или массовую скорость компонента, которая может быть определена. Минимально определяемой концентрацией является такая концентрация, для которой сигнал детектора равен удвоенной величине шума. Если уровень шума 4 мкВ, то минимально определяемой концентрации будет соответствовать сигнал детектора, равный 8 мкВ. В конечном счете уровень шумов детектора определяет нижний предел чувствительности; поэтому целесообразно использовать минимально определяемую концентрацию как меру чувствительности детектора вместо меры чувствительности в единицах мВ-см3/мг. Точность количественного анализа в основном определяется формой зависимости между концентрацией и сигналом детектора. Чем ближе эта зависимость к линейной, тем точнее анализ. Линейность показаний детектора находится как тангенс угла наклона кривой зависимости сигнала детектора, построенной в логарифмической шкале. Идеальный случай — наклон равен 1,00. Однако на практике, линейность пламенно-ионизационного детектора равна 0,95—0,99, Линейный динамический диапазон (ЛДД) — отношение максимальной концентрации к минимальной, между которыми находится область линейности детектора. Сигнал детектора определяется различными физико-химическими характеристиками анализируемых веществ и принципом работы детектора. Детектор по теплопроводности реагирует на разность теплопроводностей пробы и газа-носителя. Теплопроводность в свою очередь зависит от молекулярной массы, что означает изменение сигнала детектора в зависимости от молекулярной массы исследуемых веществ. В этом случае необходимо вводить поправочные коэффициенты. Сигнал пламенно-ионизационного детектора различен для веществ различных классов, а также и для одинаковых количеств различных веществ. Каждый детектор, кроме диафрагменного детектора Гугля—Мулярского, требует калибровки при определении поправочных коэффициентов, необходимых для количественного анализа. Принцип действия детекторов. Интегральные детекторы в настоящее время практически не используются, поэтому ниже описываются наиболее распространенные способы дифференциального детектирования. Подробно рассмотрим пять типов дифференциальных детекторов: I — по теплопроводности (катарометр), II — пламенно-ионизационный (ПИД), III — по электронному захвату (ЭЗД), IV — фосфорный (ФД), V— гелиевый (ГД). Существуют также и другие детекторы, основанные на различных принципах. I. Катарометр в настоящее время является очень распространенным детектором. Принцип его действия: нагретое тело теряет теплоту со скоростью, зависящей от состава окружающего его газа. В связи с этим скорость теплоотдачи определяет газовый состав. Основными процессами, при которых происходит унос теплоты, являются вынужденная конвекция и передача теплоты газовому потоку, которая зависит от теплопроводности газа. С этими двумя процессами связано более 75% общих потерь теплоты. Потери теплоты, обусловленные вынужденной конвекцией, можно свести к минимуму, если соответствующим образом расположить нить внутри камеры. В дальнейшем будем предполагать, что единственной причиной тепловых потерь служит теплопроводность в газе-носителе При использовании таких газов-носителей, как гелий или водород, превалируют потери теплоты путем передачи се от нити к газу.. Водород и гелий – носители пробы - обладают самой большой теплопроводностью. Основным элементом ячейки по теплопроводности служит металлическая нить, скрученная в спираль и расположенная внутри камеры в металлическом блоке. Нить изготавливается из материала, имеющего высокий температурный коэффициент сопротивления (платины, вольфрама). Нить нагревают, пропуская через нее постоянный ток. Температура нити определяется равновесием, устанавливающимся между входной электрической мощностью и мощностью тепловых потерь, связанных с отводом теплоты окружающим газом. В том случае, когда через ячейку протекает чистый газ-носитель, потери теплоты постоянны, и поэтому температура нити также постоянна. В случае изменения газового состава — при наличии анализируемого вещества — изменяется температура нити, что вызывает соответствующее изменение электрического сопротивления, которое измеряется и преобразовывается в выходной сигнал. На чувствительность катарометра оказывают влияние сила тока, газ-носитель и температура. Газ-носитель необходимо выбирать с максимально возможной теплопроводностью. Для органических соединений наиболее высокая чувствительность детектирования достигается при применении в качестве газа-носителя водорода или гелия. При увеличении силы тока чувствительность растет (при росте силы тока в два раза, чувствительность возрастает в 4—8 раз). Однако следует учитывать, что слишком сильное увеличение тока может привести к перегоранию нити и нестабильности нулевой линии. Температура нити должна быть достаточно высокой, чтобы избежать конденсации пробы внутри детектора. Несмотря на это следует все же стараться поддерживать, если это возможно, более низкую температуру детектора. Учитывая все вышеотмеченное, можно сделать следующий вывод: для повышения чувствительности катарометра необходимы увеличение силы тока в нити, уменьшение температуры блока и выбор газа - носителя с высокой теплопроводностью.   Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  ЧТО ПРОИСХОДИТ, КОГДА МЫ ССОРИМСЯ Не понимая различий, существующих между мужчинами и женщинами, очень легко довести дело до ссоры...  Что способствует осуществлению желаний? Стопроцентная, непоколебимая уверенность в своем... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|