
|
|
Классификация наследственных заболеваний н.с.1). По уровню поражения наследственного аппарата: хромосомные заболевания - генные (или молекулярные) болезни: этот подход важен для определения методов диагностики. Так, при хромосомной патологии единственным методом обследования является кариотипирование, в то же время генные болезни по хромосомному анализу диагностировать невозможно, необходимы другие методы исследования. 2). Пo клинической картине заболевания, исходя из уровня поражения нервной системы и ведущего синдрома.. Так, по этому принципу выделяются первичные мышечные дистрофии, вторичные (неврогенные и спинальные) амиотрофии, мозжечковые, пирамидные, экстрапирамидные и корковые дегенерации. 3). По биохимическому дефекту. Изучены наследственные заболевания с поражением углеводного обмена (сахарный диабет), обмена аминокислот (фенилкетонурия, гистидинемия и др.), обмена жиров (нейролипидозы), минерального обмена (гепатоцеребральные дегенерации). Однако при многих наследственных заболеваниях патогенез и биохимический дефект не известен и, естественно, большое количество наследственных заболеваний не может быть классифицировано по данному признаку. 4) По типу наследования: а) Аутосомно-доминантный тип наследования: хорея Гентингтона, болезнь Паркинсона, атаксия Пьера-Мари; б) Аутосомно-рецессивный тип наследования: болезнь Фридрейха, фенилкетонурия, спинальные амиотрофии; в) Рецессивный, сцепленный с полом, тип наследования: гемофилия, миопатия 12 Дюшенна. Менее изучены другие типы наследования: кодоминантный; доминантный, сцепленный с Х-хромосомой. Клиническая классификация наследственных болезней нервной системы по С.Н.Давиденкову. I. Прогрессирующие заболевания мышечного аппарата: 1.Миопатия: 2.Митония (б.Томсена) 3.Парамиотония Эйленбурга 4.Атрофическая миотония 5.Пароксизмальный семейный паралич II. Болезни с преимущественным поражением периферического двигательного нейрона: 1.Детская спинальная амиотрофия Верднига-Гоффмана 2.Юношеская спинальная амиотрофия Кугельберга-Веландера 3.Невральная амиотрофия Шарко-Мари-Тус 4.Гипертрофический неврит Дежерин-Сотта 5.Боковой амиотрофический склероз III. Болезни с преимущественным поражением пирамидной системы: 1.Сиастический паралич Штрумпеля 2.Семейный спастический паралич с амиотрофией, олигофренией и дегенерацией сетчатки 3.Семейный спастический паралич с ихтиозом и олигофренией IV. Болезни с преимущественным поражением аппарата координации (наследственные атаксии): 1.Спинальная атаксия Фридриха 2.Синдром Руси-Леви 3.Наследственная мозжечковая атаксия Пьера-Мари 4.Оливо-понто-церебеллярная атрофия 5.Поздняя системная атрофия коры мозжечка 6.Синдром Маринино-Сыгрена 7.Атаксия-телеангиоэктазия 8.Болезнь Рефзума V. Болезни с преимущественным поражением экстрапирамидной системы: 1.Хорея Гентингтона 2.Двойной атетоз 3.Торсионная дистония 4.б-нь Паркинсона (др.паралич) 5.Наследственное дрож. 6.б-нь Голлевордена-Шпатца 7.Гепато-лентикулярная дегенерация 8.Миоклонус-эпилепсия 9.б-нь Фара VI. Заболевания ЦНС, связанные с нарушением липидного обмена: 1.Липидозы (б-нь Гей-Сакса, Гоше) 2.Наследственные лейкодистрофии (б-нь Краббе) VII. Факоматозы – б-нь Реклингаузена, б-нь Гиппело-Линдау, б-нь Штюрге-Вебера. VII. Сирингомиелия. 103.Нервно-мышечные заболевания. Прогрессирующая мышечная дистрофия (глава 29)- большая группа болезней, при которых отмечается поражение мышечной ткани, нервно-мышечных синапсов, периферических нервов, передних рогов спинного мозга. Наиболее распространенными нервно-мышечными заболеваниями являются миопатии, миотонии и миастения. 1) МИОПАТИИ -сборная группа болезней,различных по этиологии.Всн миопатии сопровождаются мышечными поражениями,которые носят первичный (первично-мышечный)или вторичный (неврогенный) характер. § Этиология - обусловлены изменениями генетической информации и передаются по наследству (чаще по рецессивному типу) § Классификация. 1. Первичные миопатии (прогрессирующие мышечные дистрофии). 2. Вторичные миопатии (невральные и спинальные амиотрофии). 3. Смешанные формы миопатий. 4. Миопатические синдромы. § Патогенез. - первичные миопатии( усиленный распад мышечных белков и замещение их жировой и соединительной тканью) ???Патогенез неврогенных (невральных и спинальных) амиотрофий до настоящего времени не изучен. - Невральные амиотрофии(следствие генетически обусловленного поражения периферических нервов, в которых развиваются аксональная атрофия и сегментарная демиелинизация. Поражение мышц вторично и обусловлено их денервацией) - Спинальные амиотрофии(вызваны первичной дегенерацией тел мотонейронов спинного мозга и (или) их аналогов – двигательных ядер черепных нервов-нарушение функции мышц также вторично) Клиническая картина - первичная миопатия, к ней относятся: 1) детская псевдогипертрофическая миопатия Дюшенна быстро прогрессирующая, тип наследования рецессивный, сцепленный с Х-хромосомой, ген кодирует синтез белка, получившего название дистрофин(обеспечивает связь между сократительным аппаратом мышечного волокна и сарколеммой), начинается до 5 лет,умирают до 20 лет, характеризуется: нарастающей мышечной слабостью и атрофиями(с мышц тазового пояса, бедер и переходящими на мышцы плечевого пояса),что сопровождается образованием контрактур, гиперлордоз поясничного отдела, «утиная» походка, «крыловидные» лопатки, псевдогипертрофии икроножных мышц, поражаение сердечной мышцы, ожирение диспластического характера, отставания в психическом развитии. 2) Ювенильная конечностно-поясная миопатия Эрба-Рота Первые признаки проявляются на втором десятилетии жизни, клиника схожа с миопатией Дюшена. сухожильные рефлексы угасают, вставание из положения сидя «лесенкой» помогая руками, далее полная обездвиженность. 3) Плечелопаточно-лицевая миопатия Ландузи – Дежерина Значительное поражение мышц лица и плечевого пояса, протекает относительно доброкачественно. Диагностика (для первичной миопатии) клинические проявления, генеалогический анамнез, биохимические показатели,результаты гистологического и гистохимического исследования материала мышц и нервов, данные электромиографического и молекулярногенетического исследований, увеличение активности креатинфосфокиназы и альдолазы в сыворотке крови, а также низкий уровень лимонной кислоты, креатинурия и уменьшение экскреции с мочой креатинина, электромиографическое исследование, ультразвуковой и компьютернотомографический методы исследования мышц(уплотнения в мышцах, уменьшенный объем мышц, а также избыточное развитие жировой и соединительной ткани) Лечение. Курсы лечения для замедления патологического процесса или стабилизации. При первичных миопатиях: препараты, направленные на улучшение трофики мышц(ретаболил, церебролизин, витамин Е, аденозинтрифосфорную кислоту, оротат калия, рибоксин, глутаминовую кислоту) и проводимости импульсов по нервным стволам и через мионевральные синапсы(антихолинэстеразные средства (прозерин, калимин) +физиотерапия, массаж и лечебная физкультура 2) Миотонии – наследственно-дегенеративные нервно-мышечные заболевания, объединенные наличием общего симптома – миотонического феномена. Он заключается в резком затруднении расслабления мышц после сильного сокращения. Различают врожденную миотонию (болезнь Томсена), атрофическую миотонию (болезнь Россолимо – Баттена– Штейнерта – Куршманна), холодовую парамиотонию Эйленбурга и ряд других более редких нозологических форм. 3) Миастения – аутоиммунное заболевание, сопровождающееся слабостью и патологической утомляемостью мышц вследствие блокады аутоантителами постсинаптических ацетилхолиновых рецепторов нервно-мышечных соединений.   ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  Живите по правилу: МАЛО ЛИ ЧТО НА СВЕТЕ СУЩЕСТВУЕТ? Я неслучайно подчеркиваю, что место в голове ограничено, а информации вокруг много, и что ваше право... 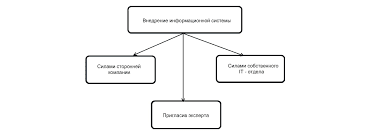 Что делает отдел по эксплуатации и сопровождению ИС? Отвечает за сохранность данных (расписания копирования, копирование и пр.)...  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|