
|
|
Роль компьютерного моделирования в материаловедении и физике конденсированных сред. Уровни описания структуры конденсированных сред.Стр 1 из 10Следующая ⇒ Роль компьютерного моделирования в материаловедении и физике конденсированных сред. Уровни описания структуры конденсированных сред. Компьютерное моделирование(КМ)- создание и проведение расчетов математических моделей на ЭВМ. КМ применяется для решения следующих задач: 1) проектирование,дизайн новых материалов; 2) описание и изучение структуры материалов; 3) описание и изучение физико-химических свойств материалов; 4) решение производственных и промышленных задач,получение новых материалов: а) конструирование и проектирование технологических процессов производства новых материалов; б) моделирование технологических процессов; в) моделирование производственных процессов; г) разработка автоматизированных производств. Уровни описания математических и компьютерных моделей: 1) макроуровень – происходит моделирование макроскопических параметров процессов и объектов; 2) мезоуровень - промежуточный уровень; 3) микроуровень включет в себя описание и моделирование структуры материалов; 4) атомный уровень. Для каждого уровня моделирования разработаны свои математические модели, методы их решения и программные системы моделирования.
Характеристика и классификация методов компьютерного моделирования в материаловедении. Моделирование из первых принципов. Атомное моделирование. По уровню описания математических и компьютерных моделей: 1) макроуровень – происходит моделирование макроскопических параметров процессов и объектов; 2) мезоуровень - промежуточный уровень; 3) микроуровень включет в себя описание и моделирование структуры материалов; 4) атомный уровень. Для каждого уровня моделирования разработаны свои математические модели, методы их решения и программные системы моделирования. По методам математического моделирования: 1)классические математические методы механики сплошных сред; 2)статистические методы и теория вероятности; 3)кванто-механические методы. Для нано- и атомного уровня в КМ математической модели наноматериалов и наносистем существуют следующие этапы: 1)Задание исходной структуры атомов; 2)Выбор потенциала межатомного взаимодействия; 3)Запись уравнений динамики или статики для выбранной структуры атомов; 4)Выбор методов решения уравнений динамики или статики атомов; 5)Выбор методов вычисления микро- и макропараметров; 6)Выбор и разработка программной системы. Создание программного кода; 7)Визуализация методов моделирования; 8)Отладка программы,проведение расчетов,получение результатов, их анализ и выводы; Современные потенциалы, учитывающие многочастичный характер взаимодействия атомов, строятся из квантовой теории электронной структуры кристаллов, или, как говорят, из первых принципов (ab initio).Эта теория основана на теории функционала плотности многоэлектронных систем, основные элементы которой мы сейчас рассмотрим. Достоинством расчётов из первых принципов является точное описание атомного взаимодействия с учётом квантовых эффектов. Недостатком — невозможность расчёта за разумное время систем с достаточно большим числом атомов (на практике редко более 100). Если расположить современные методы моделирования, по возрастанию размеров моделируемых систем и времени моделирования, то картина получится следующей: 1. Ab initio методы, не использующие приближений 2. Ab initio методы, использующие приближения 3. Методы молекулярной динамики, использующие полуэмпирические потенциалы 4. Метод Монте-Карло 5. Методы конечных элементов Аналогично от 1-5 увеличивается количество упрощений и приближений которые могут влиять на корректность получаемого результата.
Парные потенциалы Наиболее простым видом межатомных потенциалов являются парные потенциалы. Данные потенциалы являются фенологическими (экспериментально обоснованными). То есть они являются аппроксимированными. Аппроксимирующая функция для межатомного потенциала подбирается так, чтобы удовлетворять макроскопическим параметрам (коэффициенту сжатия, сублимации и т.д.)
где Наиболее распространенными парными потенциалами являются потенциалы Леннарда-Джонса и Морзе. Рис. 2. Графики функций, характеризующих потенциал МПА для Ni
а б Рис. 3. Зависимости энергии связи на один атом от параметра решетки в кристалле (а) и расстояния между атомами в димере (б) никеля Метод минимизации энергии. Построенная для моделирования атомная система в исходном состоянии, как правило, имеет конфигурацию, далеко не соответствующую минимуму потенциальной энергии взаимодействия атомов. Если эту систему предоставить самой себе, она будет релаксировать к состоянию с минимумом энергии. Во многих исследованиях, направленных, например, не на выяснение динамического поведения, а на определение структуры дефектов, нахождение такого состояния является основной задачей. Такая задача, например, возникает, при исследовании структуры точечных дефектов или дислокаций или границ зерен. Метод компьютерного моделирования, реализующий нахождение конфигурации атомной системы, соответствующей минимуму потенциальной энергии, называется методом минимизации энергии или, очень часто, применительно к атомному моделированию он называется молекулярной статикой. Физически структура, которая определяется методом минимизации энергии, представляет собой равновесную структуру, которую имеет атомная система при температуре Т = 0 К. Математически задача нахождения равновесной структуры состоит в минимизации полной потенциальной энергии взаимодействия системы Таких алгоритмов имеется много. Практически все они основаны на расчете градиента минимизируемой функции (в данном случае Одним из широко используемых в программах атомного компьютерного моделирования методов является метод сопряженных градиентов.
Рис. 4. Схематический график функции многих переменных с локальными и глобальным минимумами Наиболее трудной проблемой при минимизации энергии является нахождение глобального минимума, то есть состояния с действительно наименьшей энергией, которая возможна среди всех конфигураций. Сложные многомерные функции имеют множество локальных минимумов (см. рис. 4), и тот из них, в котором функция обладает наименьшим значением, называется глобальным. Если исходная конфигурация оказалась вблизи одного из локальных минимумов, не совпадающих с глобальным, то релаксация любым из методов приводит к этому минимуму. Для атомной системы такое состояние является метастабильным, из которого при нулевой температуре она никогда не выйдет. Таким образом, стабильная структура с минимальной энергией оказывается доступной не из любого начального состояния. Найдя то или иное метастабильное состояние, никогда нельзя определить, является ли оно стабильным, если не перебраны все возможные локальные минимумы. Более того, нет способов определения, сколько локальных минимумов существует для данной системы. Для решения этой проблемы не существует общих методов. Частично преодолеть ее помогает использование метода молекулярной динамики (МД). Ниже, при изучении методов моделирования границ зерен, мы ознакомимся с несколькими методами, которые позволяют преодолевать проблему нахождения глобального минимума именно для этих дефектов.
Граничные условия. Для моделирования поведения макроскопических систем или дефектов в макросистемах необходимо накладывать специальные условия на атомы на границе моделируемой системы, называемые граничными условиями. Существует несколько типов граничных условий. Наиболее часто используемыми являются периодические граничные условия (ПГУ). Суть этих условий заключается в следующем. Частицы заключаются в ячейке в форме параллелепипеда, называемой расчетной ячейкой. Эта ячейка повторяется бесконечное число раз путем трансляции во всех трех направлениях, заполняя все пространство (см. рис. 5). Иными словами, если одна частица расположена с координатами
Если бы потенциал имел бесконечную область действия, применение ПГУ привело бы к бесконечному количеству взаимодействующих пар. На практике, однако, ограниченный радиус действия потенциалов обеспечивает конечность числа пар. С другой стороны, использование ПГУ вносит дополнительную сложность в компьютерные программы, и эта сложность преодолевается с помощью так называемого правила ближайшей частицы: из всех возможных образов частицы j мы оставляем только ближайший, выбрасывая все остальные. Только ближайшая частица является кандидатом для взаимодействия, все остальные точно не взаимодействуют.
Это и есть алгоритм Верле.
Задание начальных положений и скоростей. Понимание статистических законов и умение пользоваться ими является ключевым моментом при изучении молекулярной физики. К сожалению, формула Максвелла распределения молекул по скоростям и по проекциям скоростей и формула Эйнштейна для броуновского движения в школьном учебнике либо не даются вовсе, либо представлены без вывода. Формулы
Распределение по скоростям для двумерного газа:
Распределение по скоростям для трехмерного газa:
Описание физической модели
Классическая статистика
, где δ — дельта-функция, а постоянная g — плотность состояний (то есть фазового объёма), определяемая условием нормировки функции распределения на единицу при интегрировании по всем различным микросостояниям:
dГ — элемент фазового объёма, который в классическом случае равен Интервал энергии
Здесь нормировочным множителем
выступает статистический вес (то есть число состояний в слое, его фазовый объём), определяемый заданными параметрами макросостояния. Квантовая статистика В квантовых системах ΔE обусловлено соотношением неопределённостей в связи со временем наблюдения. При этом можно рассматривать ансамбль полностью изолированных систем, когда ΔE/E → 0. Равномерное распределение вероятностей
Термодинамика Термодинамические потенциалы, а с ними и вся термодинамика микроканонического ансамбля строится из энтропии, напрямую связанной со статистическим весом формулойБольцмана:
, где k — постоянная Больцмана. Микроканоническое распределение неудобно здесь для практического применения, так как для вычисления статистического веса необходимо вычислить все микросостояния системы. Численное моделирование
Распределение Гиббса
Классический случай
Использование команд. С помощью команд RasMol, вводимых в командном окне, можно выполнить значительно большее количество манипуляций изображением, чем с использованием меню графического окна и мыши. Команды вводятся с клавиатуры так же, как команды DOS. При этом RasMol сохраняет предыдущие команды. Команда Ctrl+P вызывает из памяти предыдущую команду, а Ctrl+N – следующую. Эти команды позволяют выбрать формат представления изображения и записывать его в файл. Размеры и расстояния в RasMol могут быть заданы в ангстремах или единицах RasMol. Если значение какой-либо величины содержит десятичную точку, то оно считается заданным в ангстремах. Например, координаты, введенные в PDB файле с использованием десятичной точки, имеют автоматически размерность ангстрема. Единица RasMol составляет 1/250-ю часть ангстрема. Любая величина, заданная в виде целых числе, воспринимается в этих единицах. Например, команда «spacefill 300» задает сферы радиусом 300 единиц RasMol, то есть 1,2 Å. Команды программы RasMol: «Backbone Background Bond Cartoon Centre Clipboard Colour Connect CPK CPKnew Define Depth Dots Echo English Exit French HBonds Help Italian Label Load Molecule Monitor Pause Print Quit Refresh Renumber Reset Restrict Ribbons Rotate Save Script Select Set Show Slab Source Spacefill Spanish SSBonds Star Stereo Strands Structure Surface Trace Translate UnBond Wireframe Write Zap Zoom». Команды: Background Формат: background <colour> (здесь и далее в угловые скобки заключаются выражения или слова, набираемые по выбору пользователя). Задает цвет фона в графическом окне. Colour Формат: ‘colour <colour>’. Команда позволяет выбирать цвет представления выбранного типа атомов (о команде выбора атомов одного сорта ‘select’ см. ниже). Цвета атомов выбираются так же, как для фона. Rotate Команда ‘rotate <axis> <value>’ позволяет поворачивать изображение вокруг выбранной оси на выбранный угол. Параметр <axis> может иметь значения ‘x’, ‘y’ и ‘z’. Параметр <value> имеет целочисленное значение, равное углу поворота в градусах. Select Формат: ‘select <expression>’. Позволяет выбирать определенные атомы в системе. Дальнейшие команды, определяющие, например, цвет, действуют только на выбранные атомы. Выражение <expression> может представлять, например, название элемента, атомы которого выбираются (Al, Cu и т.д.). Иногда типы атомов в PDB файле определяются цифрами; тогда в <expression> используется соответствующая цифра. Примеры: ‘select Al’, ‘select 1’. Spacefill Формат: ‘spacefill <expression>’. Эта команда представляет выбранные атомы в виде сплошных сфер с размером, определяемым выражением <expression>. Если значение в этом выражении задано целым числом, единицей измерения является единица RasMol, равная 1/250-й части ангстрема. Если число задано с десятичной точкой, размер определяется в ангстремах. Размер атома не должен превышать 500 единиц (2.0 Å); при превышении этого значения программа выдает ошибку "Parameter value too large" (значение параметра слишком велико) в командной строке Zoom Формат: ‘zoom <value>’. Изменяет увеличение изображения. Максимальное значение 1562. Использование команд ‘zoom on’ и ‘zoom off’ позволяет устанавливать и отменять фиксированное увеличение. Write Формат: ‘write {<format>} <filename>. Записывает изображение в одном из стандартных графических форматов, заданных выражением <format>, в файл с названием <filename>. Поддерживаются многие распространенные графические форматы, как BMP, GIF, PS, EPSF, VECTPS (векторный пост-скрипт) и др.
Использование команд. С помощью команд RasMol, вводимых в командном окне, можно выполнить значительно большее количество манипуляций изображением, чем с использованием меню графического окна и мыши. Команды вводятся с клавиатуры так же, как команды DOS. При этом RasMol сохраняет предыдущие команды. Команда Ctrl+P вызывает из памяти предыдущую команду, а Ctrl+N – следующую. Эти команды позволяют выбрать формат представления изображения и записывать его в файл. Размеры и расстояния в RasMol могут быть заданы в ангстремах или единицах RasMol. Если значение какой-либо величины содержит десятичную точку, то оно считается заданным в ангстремах. Например, координаты, введенные в PDB файле с использованием десятичной точки, имеют автоматически размерность ангстрема. Единица RasMol составляет 1/250-ю часть ангстрема. Любая величина, заданная в виде целых числе, воспринимается в этих единицах. Например, команда «spacefill 300» задает сферы радиусом 300 единиц RasMol, то есть 1,2 Å. Команды программы RasMol: «Backbone Background Bond Cartoon Centre Clipboard Colour Connect CPK CPKnew Define Depth Dots Echo English Exit French HBonds Help Italian Label Load Molecule Monitor Pause Print Quit Refresh Renumber Reset Restrict Ribbons Rotate Save Script Select Set Show Slab Source Spacefill Spanish SSBonds Star Stereo Strands Structure Surface Trace Translate UnBond Wireframe Write Zap Zoom». Подробно о них можно узнать из руководства к программе. Команды редактирования: Background Формат: background <colour> (здесь и далее в угловые скобки заключаются выражения или слова, набираемые по выбору пользователя). Задает цвет фона в графическом окне. Colour Формат: ‘colour <colour>’. Команда позволяет выбирать цвет представления выбранного типа атомов (о команде выбора атомов одного сорта ‘select’ см. ниже). Цвета атомов выбираются так же, как для фона. Rotate Команда ‘rotate <axis> <value>’ позволяет поворачивать изображение вокруг выбранной оси на выбранный угол. Параметр <axis> может иметь значения ‘x’, ‘y’ и ‘z’. Параметр <value> имеет целочисленное значение, равное углу поворота в градусах. Select Формат: ‘select <expression>’. Позволяет выбирать определенные атомы в системе. Дальнейшие команды, определяющие, например, цвет, действуют только на выбранные атомы. Выражение <expression> может представлять, например, название элемента, атомы которого выбираются (Al, Cu и т.д.). Иногда типы атомов в PDB файле определяются цифрами; тогда в <expression> используется соответствующая цифра. Примеры: ‘select Al’, ‘select 1’.
Spacefill Формат: ‘spacefill <expression>’. Эта команда представляет выбранные атомы в виде сплошных сфер с размером, определяемым выражением <expression>. Если значение в этом выражении задано целым числом, единицей измерения является единица RasMol, равная 1/250-й части ангстрема. Если число задано с десятичной точкой, размер определяется в ангстремах. Размер атома не должен превышать 500 единиц (2.0 Å); при превышении этого значения программа выдает ошибку "Parameter value too large" (значение параметра слишком велико) в командной строке Zoom Формат: ‘zoom <value>’. Изменяет увеличение изображения. Максимальное значение 1562. Использование команд ‘zoom on’ и ‘zoom off’ позволяет устанавливать и отменять фиксированное увеличение. Write Формат: ‘write {<format>} <filename>. Записывает изображение в одном из стандартных графических форматов, заданных выражением <format>, в файл с названием <filename>. Поддерживаются многие распространенные графические форматы, как BMP, GIF, PS, EPSF, VECTPS (векторный пост-скрипт) и др. Построение карт поля упругих напряжений дислокации Программа RasMol может быть использована для построения карт напряжений по результатам расчета с помощью атомного моделирования. Большинство программ моделирования позволяет рассчитывать все 6 независимых компонентов локальных напряжений, действующих на каждый атом. Для построения карты напряжений весь интервал рассчитанных значений напряжений делится на некоторое количество n подинтервалов: (
BSAVE nskip file При моделировании с командой CMD указывает программе сохранять каждые nskip шагов в файле с названием file размеров расчетной ячейки. В каждой строке записываются 4 числа: номер шага и три размера ячейки по осям x, y, z. Использование этой команды удобно для мониторинга размеров ячейки при моделировании с динамическими границами, то есть при постоянном давлении.
ESAVE nskip file Эта команда дает указание каждые nskip шагов МД или релаксации записывать в файл с названием file значений энергии. Сохраняется средняя энергия, приходящаяся на один атом системы в единице, установленной командой EUNIT. Каждый вывод осуществляется в одну строку, в которой записываются 4 числа: номер шага, полная энергия, потенциальная и кинетическая энергия.
WRITE Команда WRITE – наиболее универсальная команда для вывода информации при моделировании. Эта команда имеет многочисленные опции, указывающие выводимые величины и названия файлов, а также режимы вывода. Один из наиболее распространенных форматов этой команды следующий: WRITE FILE [+]filename QUANTITY Здесь QUANTITY – название выводимой величины. Эта величина будет записана в файл с названием filename. Перед названием файла может быть поставлен знак “+”; тогда запись будет производиться начиная с конца этого файла. В противном случае все содержимое файл будет заменено записываемой информацией.
Рассмотрим некоторые важные значения опции OUANTITY. OUANTITY=BOX. Команда WRITE FILE filename BOX записывает размеры расчетной ячейки. OUANTITY=EATOM. Записывает номера и значения энергий всех атомов системы. OUANTITY=STRESS. Произведения компонент тензора внутренних напряжений, усредненных по всем атомам системы, на атомный объем. Единица измерения – Эрг×см. Напряжение определяется делением этого произведения на атомный объем. OUANTITY=PARTICLE. Записывает типы и координаты атомов системы. Координаты выводятся в ангстремах. Удобна для сохранения релаксированной конфигурации системы, если используется режим моделирования QUENCH. OUANTITY=POSVEL. Сохраняет типы, координаты (в ангстремах) и скорости (в см/с) всех атомов системы. Команда удобна для сохранения промежуточных или конечных состояний системы при ненулевых температурах, моделируемых командой CMD.
В XMD предусмотрена также возможность сохранения всей текущей информации, с использованием которой моделирование может быть продолжено с прерванного места. Это делается с помощью команды WRITE STATE file. Ранее сохраненное состояние системы считывается с помощью команды STATE file. В командном окне DOS выполнение программы XMD может быть прервано нажатием клавиш “CTRL+C”. При поступлении сигнала на прерывание прежде всего создается файл состояния системы на момент прерывания, которому дается название stop.sta. В дальнейшем моделирование может быть продолжено с этого шага. Команды WRITE XMOL [+]file и WRITE PDB [+]file позволяют записывать координаты атомов в форматах XMOL и PDB, которые могут быть открыты программами визуализации, в частности, программой RasMol. Для задания значения массы атомов используется команда MASS mass, где mass – значение массы атомов в атомных единицах массы (например, при моделировании Ni используется команда MASS 58.7. Для задания шага по времени используется команда DTIME dtime, где dtime – значение шага по времени в секундах. Например, команда DTIME 5.e-15 устанавливает шаг, равный 5 фс. Можно выбрать единицу измерения энергии, которая далее будет использована при любых вводах-выводах энергетических величин: EUNIT uname, где uname=erg, joule, K, eV. Чаще всего единица энергии вводится при чтении межатомного потенциала, который может быть табулирован в любой из этих единиц. Поэтому, как правило, команда EUNIT ставится в первой строке файла с таблицей потенциала. Для чтения команд из любого другого текстового файла используется команда READ filename, где filename – название файла, содержащего набор команд. Удобно бывает выделять в отдельные файлы команды, вводящие потенциал взаимодействия и исходные координаты атомов. Тогда в командном файле xmd.in будут команды чтения из этих файлов, например, READ ni.ptf и READ ni.in, где ni.ptf – файл, содержащий описание потенциала для никеля, а ni.in – файл, содержащий координаты атомов кристаллической решетки никеля. Для многих металлов доступны уже готовые таблицы потенциалов, тогда как исходные структуры чаще всего приходится создавать самим. Для описания исходной конфигурации используются команды POSITION или PARTICLE (они являются синонимами). Формат этой команды следующий. В первой строке задается POSITION n, где n – число частиц. В последующих n строках вводятся типы и координаты всех частиц: первое число обозначает тип атома (если моделируется чистый металл, то цифра 1, если сплав двух элементов – то цифры 1 и 2), три следующие – координаты x,y,z атома в ангстремах. Например, следующие строки вводят исходную конфигурацию системы из трех атомов:
READ coord.in
coord.in:
position 3 1 0.24E-01 0.76E+02 0.88E+00 1 0.52E-01 0.16E+02 0.11E+00 1 0.34E+00 0.76E+01 0.88E+01
Особенность программы XMD заключается в том, что все частицы должны находиться в первом октанте системы координат. То есть, левый нижний угол расчетной ячейки всегда совпадает с началом координат. Для задания расчетной ячейки используется команда BOX xbox ybox zbox, где xbox ybox zbox определяют координаты правого верхнего угла расчетной ячейки в ангстремах. Как правило, параметры расчетной ячейки рассчитываются при построении исходной структуры системы, поэтому их удобно записывать в одном файле с координатами атомов. Начальная температура системы задается командой ITEMP temp, где temp – значение температуры в кельвинах. Для поддержания температуры в процессе моделирования используется команда CLAMP temp [cstep]. Значение temp задает требуемую температуру. На каждом шаге МД скорости частиц умножаются на величину (T/temp)**(1/(2*cstep)), где T – мгновенная температура на данном шаге. По умолчанию границы расчетной ячейки при моделировании в XMD фиксированы. Для моделирования при постоянном давлении используется команда PRESSURE CLAMP bulkmodulus [cstep]. При использовании этой моды стенки расчетной ячейки флуктуируют с амплитудой, обратно пропорциональной объемному модулю упругости bulkmodulus. Значение последнего задается в мегабарах (1 бар = 100 кПа). Не обязательно его указывать точно, достаточно задать приближенное значение. Параметр cstep, который может отсутствовать, определяет, как быстро флуктуирует размер ячейки в ответ на внутреннее давление. Если с помощью этой команды динамические граничные условия включены, то можно задать внешнее давление командой PRESSURE EXTERNAL pressureX [ pressure Y pressure Z ]. Можно задать только одно значение давления (в МБар), тогда будет задано изотропное давление. Задав три значения, можно определить независимые давления вдоль трех осей координат.
Программа. read fcc_in.xmd read niu3.ptf pressure clamp 2 mass 58.7 dtime 5.e-15 esave 10 rel.txt repeat 50 quench 20 1 write file +box.txt box write file energy.txt eatom write file stress.txt +stress write pdb fcc.pdb end
Роль компьютерного моделирования в материаловедении и физике конденсированных сред. Уровни описания структуры конденсированных сред. Компьютерное моделирование(КМ)- создание и проведение расчетов математических моделей на ЭВМ. КМ применяется для решения следующих задач: 1) проектирование,дизайн новых материалов; 2) описание и изучение структуры материалов; 3) описание и изучение физико-химических свойств материалов; 4) решение производственных и промышленных задач,получение новых материалов: а) конструирование и проектирование технологических процессов производства новых материалов; б) моделирование технологических процессов; в) моделирование производственных процессов; г) разработка автоматизированных производств. Уровни описания математических и компьютерных моделей: 1) макроуровень – происходит моделирование макроскопических параметров процессов и объектов; 2) мезоуровень - промежуточный уровень; 3) микроуровень включет в себя описание и моделирование структуры материалов; 4) атомный уровень. Для каждого уровня моделирования разработаны свои математические модели, методы их решения и программные системы моделирования.
  ЧТО ТАКОЕ УВЕРЕННОЕ ПОВЕДЕНИЕ В МЕЖЛИЧНОСТНЫХ ОТНОШЕНИЯХ? Исторически существует три основных модели различий, существующих между...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор...  Что вызывает тренды на фондовых и товарных рынках Объяснение теории грузового поезда Первые 17 лет моих рыночных исследований сводились к попыткам вычислить, когда этот...  Система охраняемых территорий в США Изучение особо охраняемых природных территорий(ООПТ) США представляет особый интерес по многим причинам... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|
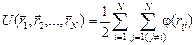 , (4)
, (4) ‑ расстояние между атомами пары i-j.
‑ расстояние между атомами пары i-j.

 , которая представляет собой сложную функцию многих переменных. Поэтому для решения этой задачи используются наиболее эффективные методы и алгоритмы, разработанные в математике для минимизации функций.
, которая представляет собой сложную функцию многих переменных. Поэтому для решения этой задачи используются наиболее эффективные методы и алгоритмы, разработанные в математике для минимизации функций. ) на каждом шаге и составлении некоторой итерационной схемы, приводящей к многомерной точке, в которой этот градиент равен нулю (необходимое условие минимума), а вторые производные положительны (достаточное условие минимума). Алгоритмы различаются именно реализацией этой итерационной схемы.
) на каждом шаге и составлении некоторой итерационной схемы, приводящей к многомерной точке, в которой этот градиент равен нулю (необходимое условие минимума), а вторые производные положительны (достаточное условие минимума). Алгоритмы различаются именно реализацией этой итерационной схемы.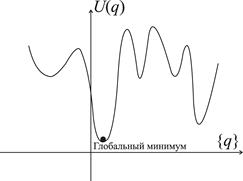
 в расчетной ячейке, считают, что эта частица в действительности представляет бесконечную совокупность частиц, расположенных в точках
в расчетной ячейке, считают, что эта частица в действительности представляет бесконечную совокупность частиц, расположенных в точках 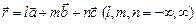 , где
, где 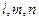 ‑ целые числа и
‑ целые числа и 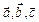 ‑ векторы, соответствующие ребрам расчетной ячейки. Все эти частицы-“изображения” движутся вместе, и только одна из них представляется в компьютерной программе. Ключевым является предположение, что каждая частица i в расчетной ячейке взаимодействует не только с остальными частицами в этой же ячейке, а также с их образами в соседних ячейках. То есть, взаимодействия могут осуществляться через границы расчетной ячейки. Это приводит к тому, что фактически (а) мы исключаем влияние поверхности на систему, и (б) положение границ ячейки относительно частиц не играет роли (то есть, трансляция ячейки по отношению к частицам не меняет сил). В процессе движения какая-либо частица системы может покинуть расчетную ячейку. В таком случае через противоположную границу ячейки в нее входит периодический образ этой частицы, так что число атомов в ячейке не изменяется.
‑ векторы, соответствующие ребрам расчетной ячейки. Все эти частицы-“изображения” движутся вместе, и только одна из них представляется в компьютерной программе. Ключевым является предположение, что каждая частица i в расчетной ячейке взаимодействует не только с остальными частицами в этой же ячейке, а также с их образами в соседних ячейках. То есть, взаимодействия могут осуществляться через границы расчетной ячейки. Это приводит к тому, что фактически (а) мы исключаем влияние поверхности на систему, и (б) положение границ ячейки относительно частиц не играет роли (то есть, трансляция ячейки по отношению к частицам не меняет сил). В процессе движения какая-либо частица системы может покинуть расчетную ячейку. В таком случае через противоположную границу ячейки в нее входит периодический образ этой частицы, так что число атомов в ячейке не изменяется.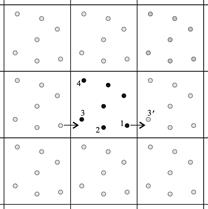
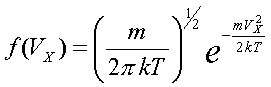
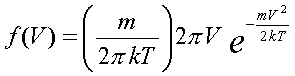
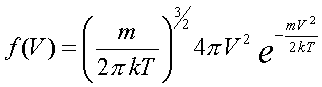

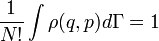
 , а в квантовом случае в трёхмере
, а в квантовом случае в трёхмере 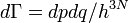 , где h — постоянная Планка.
, где h — постоянная Планка.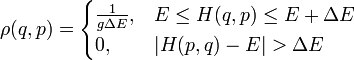
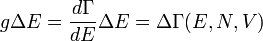
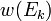 квантовых состояний с энергиями
квантовых состояний с энергиями 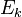 в слое (E, E + ΔE) имеет аналогичный вышеописанному вид:
в слое (E, E + ΔE) имеет аналогичный вышеописанному вид: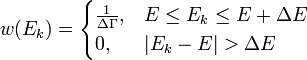
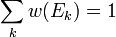
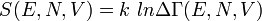
 зависит только от значения энергии и задаётся распределением Гиббса
зависит только от значения энергии и задаётся распределением Гиббса 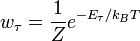 ,
, 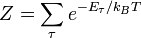 .
.  одинаковых частиц, называется статистической суммой, которая задаётся формулой.
одинаковых частиц, называется статистической суммой, которая задаётся формулой. 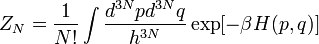
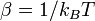 . Соответствия с общим случаем:
. Соответствия с общим случаем:  ,
, 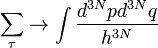 а
а 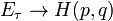 . Множитель
. Множитель  появляется в соответствии спринципом неразличимости частиц.
появляется в соответствии спринципом неразличимости частиц.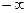 ,s1), (s1, s2),…, (s n -1, s n), (s n,
,s1), (s1, s2),…, (s n -1, s n), (s n, 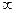 ). Атомам, напряжение на которых относится к одному и тому же подинтервалу, присваивается одно и то же название элемента, с которым они записываются в файл PDB. Затем при визуализации каждому элементу присваивается свой цвет или градация серого цвета, а размер атомов выбирается так, чтобы они заполняли все пространство без пустот. В результате каждая область, в которой напряжения относятся к одному интервалу, окажется окрашенной в один цвет.
). Атомам, напряжение на которых относится к одному и тому же подинтервалу, присваивается одно и то же название элемента, с которым они записываются в файл PDB. Затем при визуализации каждому элементу присваивается свой цвет или градация серого цвета, а размер атомов выбирается так, чтобы они заполняли все пространство без пустот. В результате каждая область, в которой напряжения относятся к одному интервалу, окажется окрашенной в один цвет.