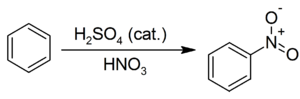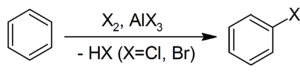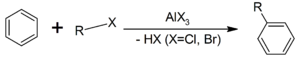|
|
Кислотность и основность органических соединений. Типы кислот Бернстеда. Факторы, определяющие кислотность органических соединений. Основания Бернстеда.Кислотность и основность – важнейшие понятия, определяющие многие физико-химические свойства и реакционную способность органических соединений. Для оценки кислотности и основности органических соединений наибольшее значение имеют две теории – теория Бренстеда и теория Льюиса. По теории Бренстеда (протолитической теории) кислотность и основность соединений связывается с переносом протона Н + Кислоты – вещества, способные отдавать протон (доноры протонов). Основания – вещества, способные присоединять протон (акцепторы протона). Кислота и основание образуют сопряженную кислотно-основную пару. Кислотные свойства проявляются в присутствии основания, основные – в присутствии кислоты. Между кислотой и основанием сопряженной пары существует взаимосвязь: чем сильнее кислота, тем слабее сопряженное основание, и наоборот. Например, уксусная кислота слабее, чем соляная, и, с Кислоты Бренстеда. Сила кислот Бренстеда зависит от растворителя. Это связано с тем, что молекулы растворителя сами являются кислотами или основаниями Бренстеда. Например, при растворении кислоты Н-А в воде происходит ионизация этих веществ: Н-А + Н2О А- + Н3О+ Концентрация воды – величина постоянная. Произведение Кр[Н2О]= Ка – константа кислотности. Чем больше величина Ка, тем сильнее кислота. Значения Ка для большинства органических веществ имеют маленькие значения, поэтому удобнее использовать отрицательный логарифм от величины Ка: рКа = - lg Ка. Чем меньше величина рКа, тем сильнее кислота. В зависимости от природы элемента, с которым связан отщепляемый протон, бренстедовские кислоты делятся на ОН-кислоты (карбоновые кислоты, спирты, фенолы), SH-кислоты (тиолы), NH-кислоты (аммиак, амины, амиды), СН-кислоты (углеводороды и их производные). По возрастанию кислотности кислоты Бренстеда располагаются в ряд: СН-кислоты < NH-кислоты < ОН-кислоты < SH-кислоты Основания Бренстеда. Основания для образования связи с протоном должны содержать либо неподеленную пару электронов, либо электроны p-связи. В соответствии с этим основания Бренстеда делятся на две группы: n-основания (содержат свободную электроную пару) и p-основания К p-основаниям относят алкены, алкадиены, арены, т.е. соединения, содержащие p-связи. p-Основания за счет электронной пары p-связи могут присоединять протон (s-орбиталь протона перекрывается с p-орбиталью). Эта неустойчивая частица носит название p-комплекса. Протон при этом не присоединяется ни к одному из атомов: В дальнейшем p-комплекс преобразуется в s-комплекс. n -Основания связь с протоном образуют за счет свободной электронной пары атома азота (в аминах, гетероциклических соединениях), атома кислорода (в спиртах, простых эфирах, карбоновых кислотах, альдегидах, кетонах), атома серы (в тиоспиртах, тиоэфирах). Сила n -оснований уменьшается в ряду: R-NH2 > R-OH > R-SH.
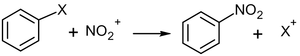
На силу кислот и оснований влияют строение вещества и природа растворителя. Кислотность органических соединений зависит: · От электроотрицательности атомов, ответственных за проявление кислотных свойств, · От поляризуемости связей в кислотном центре; · От факторов, которые определяют высокую стабильность образующихся при диссоциации. Факторы, определяющие кислотность Сила кислоты определяется стабильностью сопряженного этой кислоте основания (аниона) (чем стабильнее анион, тем сильнее кислота) Природой и зарядом атома в кислотном центре (электроотрицательностью и поляризуемостью атома, отдающего протон) Электроноакцепторные (ЭА) заместители повышают кислотность, электронодонорные (ЭД) — понижают кислотность Факторы, определяющие основность Их влияние, как правило, противоположно, влиянию факторов, определяющих кислотность 10. Реакции электрофильного замещения у ароматических соединений. Механизмы и примеры замещения. Правило ориентации при замещении в ароматических производных.. Реакции SEAr Механизм реакции SEAr или реакции ароматического электрофильного замещения (англ. Electrophilic aromatic substitution) является самым распространенным и наиболее важным среди реакций замещения ароматических соединений и состоит из двух стадий. На первом этапе происходит присоединение электрофила, на втором — отщепление электрофуга:
В ходе реакции образуется промежуточный положительно заряженный интермедиат (на рисунке — 2b). Он носит название интермедиат Уэланда, арениевый ион, арений-катион, или σ-комплекс. Этот комплекс, как правило, очень реакционноспособен и легко стабилизируется, быстро отщепляя катион. Лимитирующей стадией в подавляющем большинстве реакций SEAr является первый этап. Скорость реакции SEAr обычно представляется в следующем виде[2]: В качестве атакующей частицы обычно выступают относительно слабые электрофилы, поэтому в большинстве случаев реакция SEAr протекает под действием катализатора — кислоты Льюиса. Чаще других используются AlCl3, FeCl3, FeBr3, ZnCl2. В этом случае механизм реакции выглядит следующим образом (на примере хлорирования бензола, катализатор FeCl3)[3]: 1.На первом этапе катализатор взаимодействует с атакующей частицей с образованием активного электрофильного агента: {\displaystyle {\mathsf {Cl\!\!-\!\!Cl+FeCl_{3}}}\rightleftarrows {\mathsf {Cl\!\!-\!\!Cl^{+}}}\!\cdot \cdot \cdot {\mathsf {FeCl_{3}^{-}}}\rightleftarrows {\mathsf {Cl^{+}FeCl_{4}^{-}}}} 2. На втором этапе, собственно, и реализуется механизм SEAr:
Типичные реакции ароматического электрофильного замещения 1. Нитрование ароматических систем азотной кислотой в присутствии серной кислоты с получением нитросоединений:
Образование активной частицы[2]: {\displaystyle {\mathsf {HNO_{3}+2H_{2}SO_{4}}}\rightarrow {\mathsf {NO_{2}^{+}+H_{3}O^{+}+2HSO_{4}^{-}}}} 2. Сульфирование бензола с получением сульфокислоты:
Активной частицей в реакции является SO3. 3. Галогенирование бензола бромом, хлором или йодом приводит к образованию арилгалогенидов. Катализатором реакции выступает галогенид железа (III):
Образование активной частицы[2]: {\displaystyle {\mathsf {X_{2}+FeX_{3}}}\rightarrow {\mathsf {X^{+}+FeX_{4}^{-}}}}4. Реакция Фриделя-Крафтса — ацилирование или алкилирование с использованием ацил- или алкилгалогенидов. Типичным катализатором реакции служит хлорид алюминия, но может использоваться любая другая сильная кислота Льюиса.
Реакционная способность и ориентация в производных бензола Заместители в бензольном кольце могут как способствовать реакции замещения (активирующие заместители), так и замедлять скорость реакции (дезактвирующие заместители). Некоторые группы ориентируют замещение в орто- и пара- положения, другие — в мета. Влияние различных групп на реакционную способность объясняется устойчивостью, иначе говоря энергией активации, требующейся для получения трех возможных промежуточных интермедиатов[1]. Реакционная способность и ориентация различных групп в производных бензола[1][4]:
В замещенных бензолах возможна так называемая ипсо -атака, то есть замещение имеющегося заместителя на другой:
  ЧТО ПРОИСХОДИТ ВО ВЗРОСЛОЙ ЖИЗНИ? Если вы все еще «неправильно» связаны с матерью, вы избегаете отделения и независимого взрослого существования...  Конфликты в семейной жизни. Как это изменить? Редкий брак и взаимоотношения существуют без конфликтов и напряженности. Через это проходят все...  ЧТО И КАК ПИСАЛИ О МОДЕ В ЖУРНАЛАХ НАЧАЛА XX ВЕКА Первый номер журнала «Аполлон» за 1909 г. начинался, по сути, с программного заявления редакции журнала...  Что делать, если нет взаимности? А теперь спустимся с небес на землю. Приземлились? Продолжаем разговор... Не нашли то, что искали? Воспользуйтесь поиском гугл на сайте:
|